
It is common understanding and practice that an appropriate dissolution test is to be developed for a particular drug product when the product itself is being developed. The rationale is that the product development stage provides a sufficient variety of formulation/manufacturing differences and in vivo (bioavailability/bioequivalence) data to establish the relevance and validity of the proposed dissolution test. The choice of experimental conditions (such as apparatus, rpm, medium, pH, etc.) which fit the product’s behavior be chosen as the best or most appropriate dissolution test for future use. If one method may not fulfill the need or requirement, as commonly happens, then two or more methods for the same product may be suggested such that one (simpler) would be used as a QC test and the other (somewhat complex) as bio-relevant. In short, while products are developed, dissolution test(s) are being developed for the product evaluation.
On the other hand, one of the main uses of a dissolution test often described is to facilitate product development by establishing its drug release or dissolution characteristics. It is interesting to note that as described in the previous paragraph where a dissolution test is being chosen, the same process is also concurrently considered as the use of dissolution testing for product development. Both product and method developments are interdependent or they use each other to establish each other’s use and credibility. It is exactly like measuring something while establishing the “scale” for measuring by using the object to be measured. This practice is not only scientifically invalid but based on flawed logic as well.
Dissolution method development and product development steps must be treated as two separate steps. A dissolution method should not be used for product development until and unless it has been clearly shown independently, prior to its use, the method can determine drug dissolution (or release). A biopharmaceutic laboratory should require from an analytical chemistry laboratory or a vendor of the dissolution testers evidence that a dissolution tester (apparatus and associated experimental conditions) can determine the drug dissolution properties of products. Determining drug dissolution characteristics does not mean determining drug concentrations in different solutions at different times using multiple stirring speeds. Determining drug dissolution characteristics means that as the products are developed for human use, a formulator likes to know from an analytical laboratory how the drug is going to be released/behaved in humans. Therefore, a MANDATORY requirement for a dissolution tester is to demonstrate that it is capable of evaluating drug dissolution in humans. Unfortunately, at present, formulators/analysts both consider a dissolution tester as a validated tester if it is listed in a compendium and/or available from a vendor rather than based on its required capability of measuring drug dissolution. At present, available apparatuses lack evidence to show that they can provide drug dissolution characteristics of products.
The question is what evidence one would need to establish the validity of a tester as a dissolution tester. There are two requirements for this:(1) the tester must be capable of providing dissolution results of a product, established independently such as from pharmacokinetic studies; (2) the tester/method must be capable of differentiating dissolution characteristics of products (e.g. IR vs ER) as one would see in humans. In addition, as dissolution testing is linked to human physiology, which remains constant from product to product, these two testing conditions/criteria must be met using constant experimental conditions. Suppose a tester is not capable of providing expected/known dissolution results and not capable of differentiating products having different dissolution characteristics using common experimental conditions. In that case, the tester/method is NOT a dissolution tester/method and cannot and should not be used for product development.
At present, none of the apparatuses with associated experimental conditions appears to meet the fundamental requirement of a dissolution tester/method; thus, unfortunately, none of these testers may be used for product development and/or their evaluations. The formulators and analysts should be aware of this limitation and deficiency. On the other hand, formulators and analysts are expected to use “whatever is available”, which results in confusion and a large array of bizarre dissolution practices under different names and terminology, as shown here (Link). A huge amount of work/resources have gone into the practice of drug dissolution testing, which understandably is often rationalized with vigor, but the fact remains that these practices have been of limited or no use in determining dissolution characteristics of products. An even more frustrating aspect of the current practices of dissolution testing is that analysts and formulators are expected to develop IVIVC using testers that have never been shown for their in vivo relevance. In short, therefore, a new approach is needed to fulfill this gap of providing an appropriate dissolution tester/method.
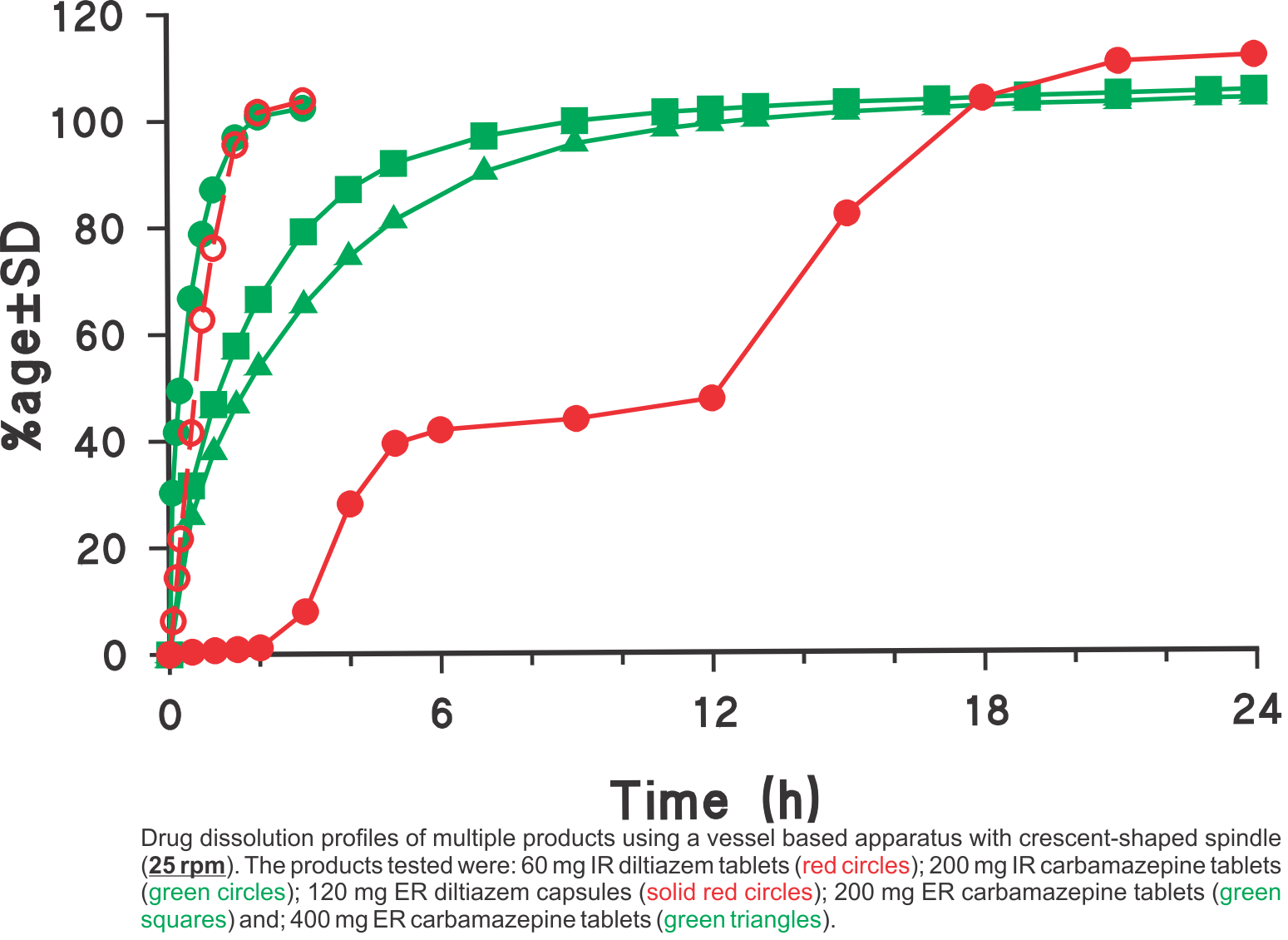
One option is to use a vessel-based dissolution tester with a modified stirrer known as “Crescent-Shaped Spindle” which has been designed by considering the deficiencies of the current practices. The spindle description and its use have been described in the literature and on this site; for example, see links [1, 2, 3].

{kind=link}