Recently FDA provided 1- and 2-pager guidance documents for chloroquine and hydroxychloroquine, respectively (link).
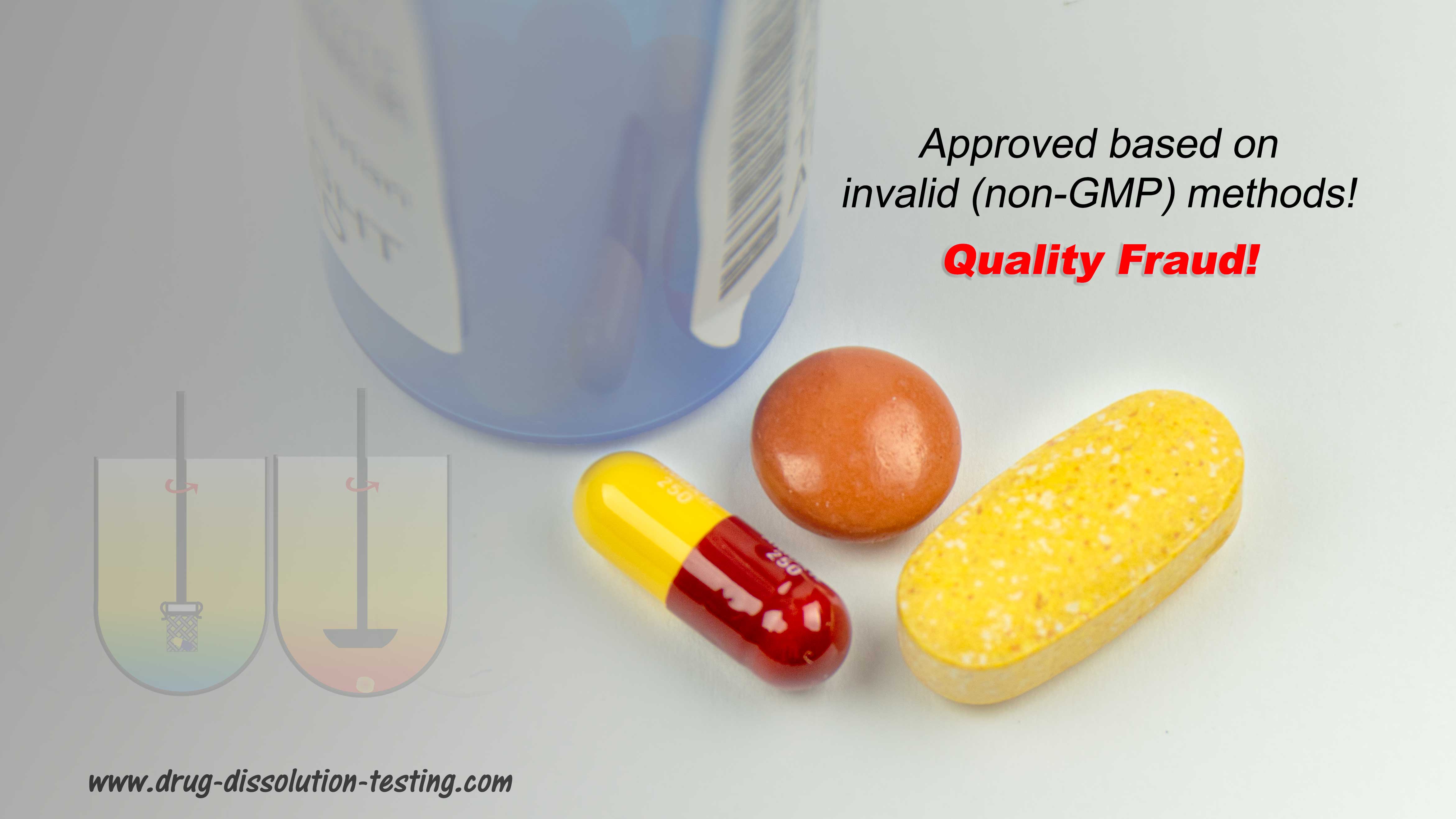
The most interesting part is that one can get product approval based on dissolution testing alone. This is what has recently been suggested in one of my recent published articles, i.e., products (“quality”) assessment can easily and accurately be established with drug dissolution testing alone (link). Therefore, there is really no need to conduct bioequivalence (BE) assessments. These (BE) assessment procedures have never been validated of the intended purpose. In fact, BE is scientifically invalid and can provide false conclusions and assurance about product quality. In addition, such testing exposes healthy human volunteers to highly potent chemicals under the disguise of medicine development.
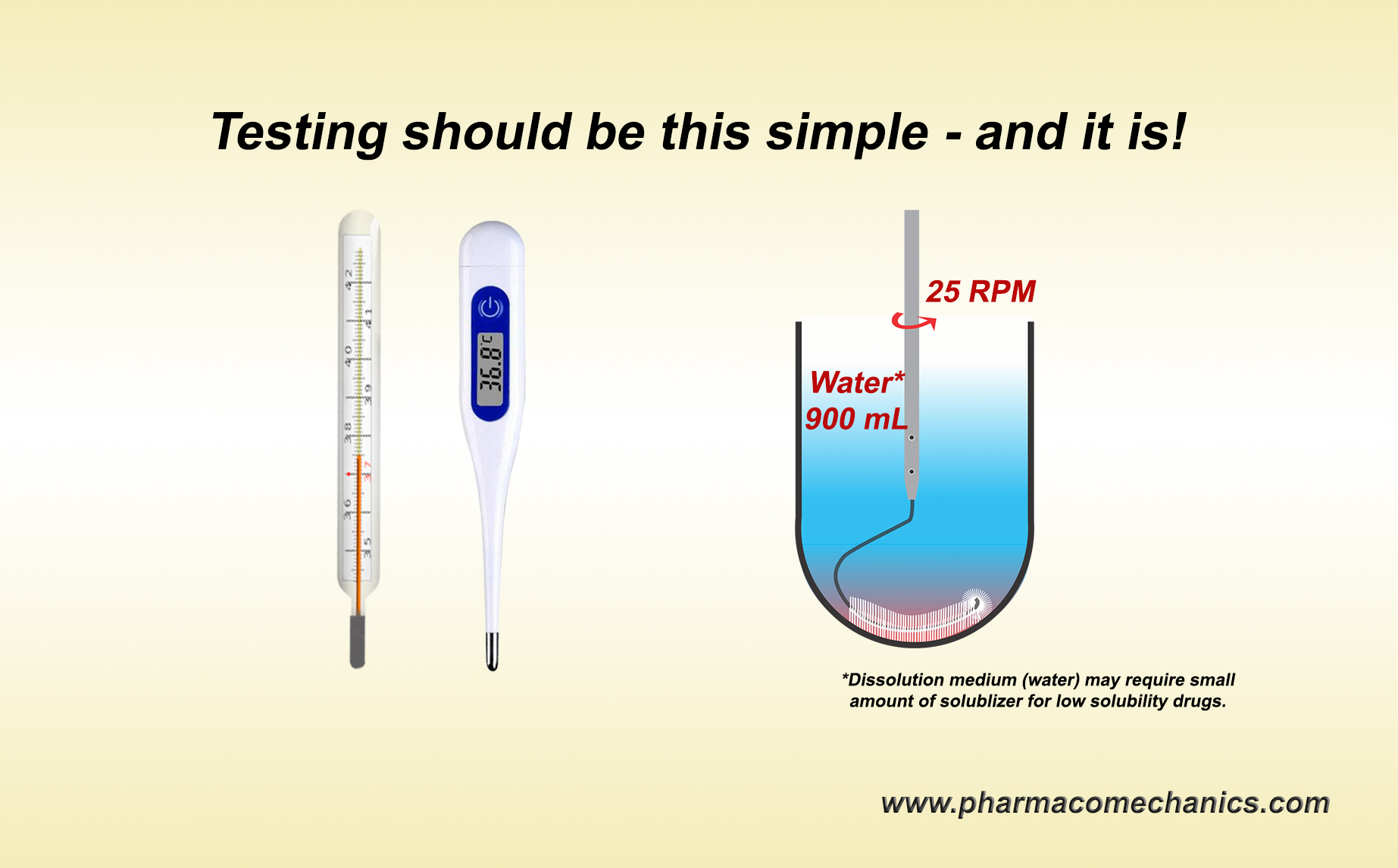
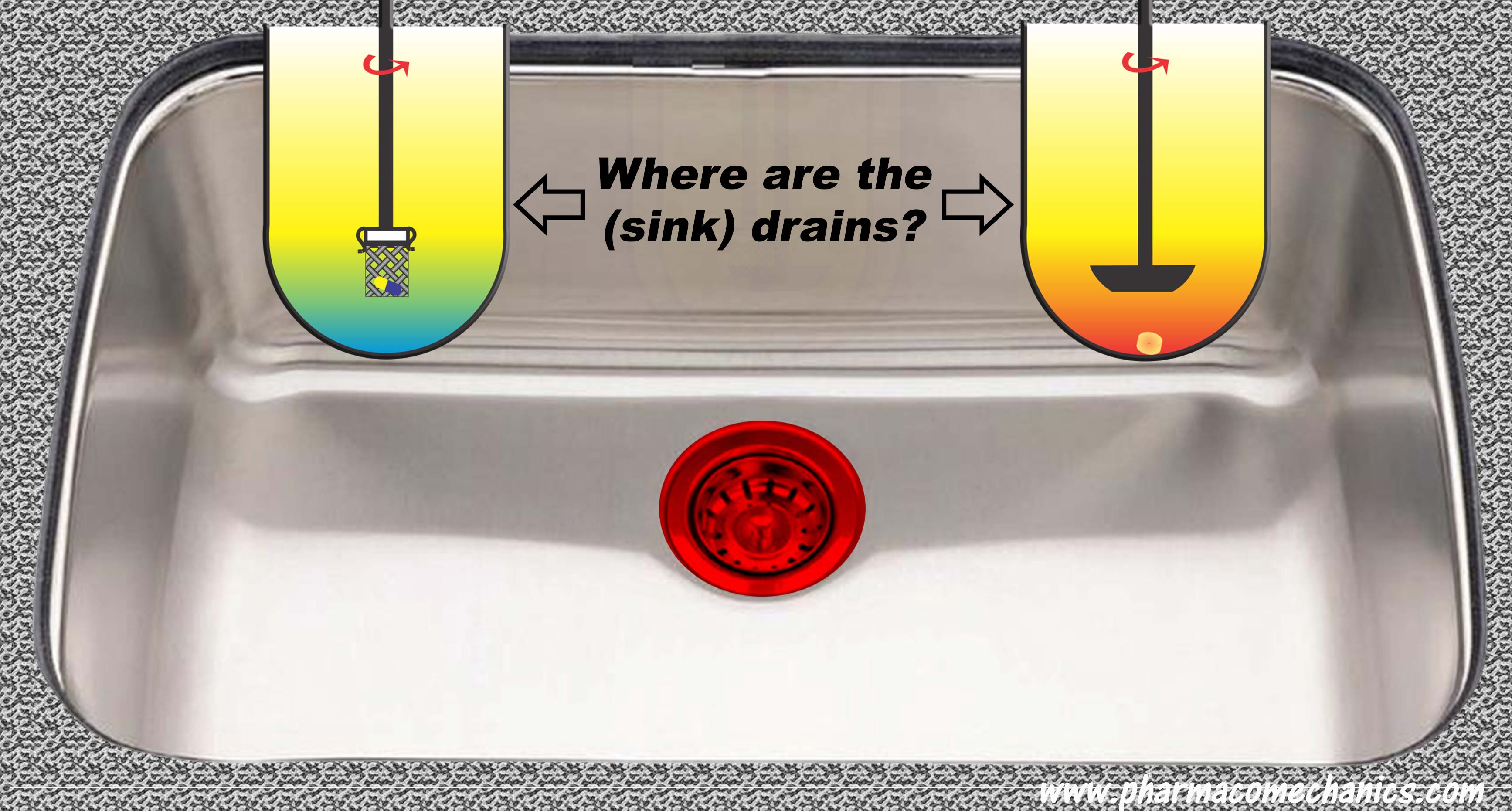
On the other hand, switching to dissolution testing alone using currently recommended USP apparatuses is not valid either, at least scientifically. The recommended apparatuses are non-GMP compliant and can provide false and irrelevant results because of their intrinsic design and operation problems. Simpler and scientifically valid options are available and could be used (link).