
The most common dissolution/release tolerances for tablet and capsule products as per USP are 80% of the drug dissolved/released at the suggested time. The question is, where does the remaining 20% of the drug go, especially, when the product meets the requirement of Assay and CU of 100%. From a consumer/patient perspective, it is like buying a 1L milk carton but being assured of receiving only 800 mL! Please, note that in many cases, the USP tolerances can be as low as 70%.

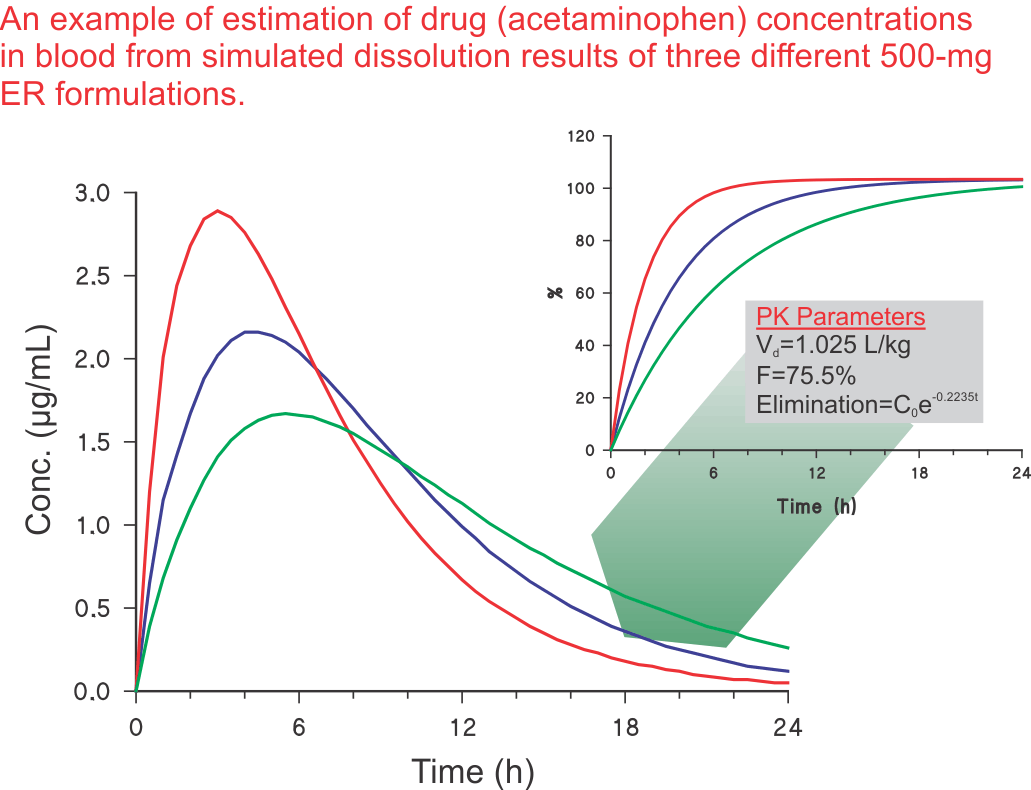
If USP tolerances are considered correct, then a product user is expected to receive less and/or an inconsistent amount of drugs. It is to be noted that all bioavailability/bioequivalence (BA/BE) studies results are reported based on 100% deliverable drug. As Assay and CU show, on average, 100% content and BA/BE assumes delivery of 100% of the drug in humans, it indicates that demonstration or requirement of less than 100% in vitro dissolution/release is an inaccurate tolerance (standard).
The reason for this discrepancy (observance of lower in vitro dissolution/release) is the poor stirring and mixing environment within the dissolution vessels using paddle and basket stirrers. The poor stirring environment creates unstirred pockets within the vessels where the drug hides. These pockets depend on the nature of the product, in particular excipients, and can hide as much as 40% of the drug as in the case of the dissolution results of the USP Performance Verification Tablets (see Figure). If this flaw of unstirred pockets is addressed, then one can observe the release/dissolution of 100% of the drug. This means that not only will dissolution results match with the Assay and CU results, but SEPARATE monitoring of Assay and CU becomes redundant. This leads to simplification, efficiency, and accuracy in overall product evaluation and development (see also related links 1, 2, 3).


{kind=link}
{kind=link}