Although not generally recognized, current practices of dissolution testing seek to adjust experimental conditions to obtain desired or expected drug release/dissolution characteristics. The variations or adjustments in experimental conditions, which are commonly referred to as method development steps, often relate to the choice of apparatuses, RPMs or flow rates, media (nature and volumes), etc. Thus, current practices defeat the purpose of dissolution testing, which is supposed to be conducted to evaluate or establish drug release characteristics of a product, in particular, at the method development stage. In addition, most standards and requirements are based on selecting and setting experimental conditions rather than evaluating products.
For appropriate dissolution testing, the tests are to be conducted using product-independent and fixed experimental conditions. This will facilitate finding the actual drug dissolution or release characteristics of a product or help in knowing one’s product. If one keeps on changing the experimental conditions, one will never know the products’ true (dissolution) characteristics.
The most widely used and referred dissolution tolerances are based on the USP Acceptance Table. The results are evaluated in stages. This means repeats are allowed with relaxed tolerances and a higher degree of variances for each subsequent test.
Stage 1:
Test 6 tablets. Each unit not less than Q+5% dissolved.
Stage 2:
Test 12 tablets (including 6 from stage 1). Average is equal or greater than Q, but no unit less than Q-15%.
Stage 3:
Test 24 tablets (including 12 from stage 1 and 2). Average equal and greater than Q, but no more than two units are less than Q-15%, and no unit less than Q-25%.
(The Q-values are provided in individual product monographs, representing expected percent drug release (dissolution) at times, such as 30, 45, 60 minutes etc.)
Considering the above criteria with a Q-value of 80, one can obtain the following set for acceptable results.
1
2
3
4
5
6
Mean
%RSD (CV)
Stage 1
95
100
91
90
98
102
96
5
Stage 2
96
88
65
110
66
65
82
23
Stage 3
55
61
92
98
105
102
85
26
103
87
77
97
93
89
91
10
Therefore, one may observe an RSD (CV) of 20% or more, and the results/product would be of acceptable quality for regulatory purposes. The expected high variability (RSD/CV) in results is built in the tolerances.
In this particular case, the test would meet the criteria at the stage 1, thus testing for the next stages would not be required. However, this may be a random phenomenon and one may get results as shown for Stage 2 or 3, at the first stage as well. These tolerances are for last dissolution sampling time, where dissolution is the highest with the least variable results. However, if dissolution results are to be reported for earlier times (such as for extended-release products), the expected variability would be higher.
Therefore, the above discussion clearly indicates that the dissolution results, particularly using Paddle/Basket apparatuses, are expected to be highly variable, often more than 20% RSD. Therefore, as often desired or suggested, setting tolerances tighter would not be scientifically valid or achievable. In that case, one would face a higher number of failures without any apparent reason, as is the case with the testing of the USP PVT prednisone tablets where tolerances are usually set tighter than one would observe in real life, thus failures.
It is a very well-established fact that the USP apparatus PVT (Performance Verification Test) using prednisone tablets faces significant criticism regarding the lack of its relevance to the performance of the apparatuses. This criticism originates from the unexpected/unpredicted failures of the PVTs, by providing (dissolution) results outside the expected ranges, also called suitability ranges. Although not generally recognized, the main reason for such failures is that expected ranges are set tighter than needed to reflect the test’s true (high) variability. Therefore, in reality, when a PVT fails, it does not reflect a substandard apparatus or the testing but is a reflection of the actual/true variability of the testing. Often suggestions are given for addressing this situation by adjusting apparatus/testing parameters. However, a common view in the scientific community is that repeating the test, single or multiple times, often provides the desired outcome. Therefore, the current practice of PVT has become an exercise of obtaining dissolution results within an expected range rather than evaluating the performance of the apparatus/test.
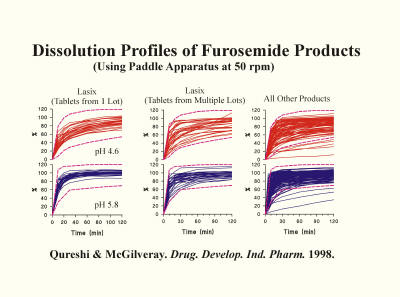
In addressing the potential cause of this high variability and unpredictability, a common focus has been the quality of the PVT (prednisone) tablets. However, if this had been true, the issue could have been resolved by using different tablets of the same or different active ingredients. In addition, there are many studies described in the literature using many other well-established products approved for human use, which demonstrate similar high variability in dissolution results. As an example, the figure below shows the dissolution characteristics of furosemide tablet products analyzed in different laboratories. The results show similar, if not worse, variability than of prednisone (PVT) tablets. Then, why is the high variability aspect often described with respect to prednisone (PVT) and not for other products? The reason is that prednisone (PVT) tablets are analyzed in multiple labs, thus higher visibility of the problem (variability). On the other hand, other products are usually tested internally, within the lab, or in few labs. Thus the problem is less visible. However, variability and unpredictability of results in both cases, PVT or products testing, is exactly the same.
The obvious logical conclusion from such observations would be that if the problem is not because of the tablets, the technique could potentially be the reason. However, studies are now reported in the literature, based on laboratory experiments and computer simulation exercises, which clearly demonstrate that the currently used apparatuses (Paddle/Basket) should indeed provide highly variable and unpredictable results. This variability and unpredictability results come from the poor flow dynamics within dissolution vessels, resulting in highly variable and unpredictable product-medium interactions. Therefore, it is a problem of the apparatuses/technique and not that of the products or PVT tablets, in particular.
Obviously, as the technique/apparatuses are the prime suspects of the high variability and unpredictability problem, then all results obtained using these apparatuses will become suspect, potentially undermining the quality of the products and adding cost to the product development and evaluation.
Often a reason is provided, in the literature, that it is a test to differentiate drug release characteristics of a product reflecting the potential impact of formulation and/or manufacturing differences. This type of test usually does not link or associate with the products’ in vivo drug release characteristics. Such tests are often referred to as “discriminatory tests.” The underlying reason for such a “discriminatory test” is that if the test relates dissolution differences to formulation/manufacturing differences, then the test will flag potential deviation from the expected behavior of the product in vivo (humans). The main assumption here is that the differences in formulation and/or manufacturing attributes would result in vivo drug release characteristics, producing a substandard product. This, unfortunately, is an incorrect assumption because the differences in formulation and/or manufacturing, on their own, do not necessarily result in differences in vivo drug release, at least not in most cases. If this assumption had been correct, then generic products and the industry would not have existed. The reason being, the generic products, which are based on vastly different formulation and manufacturing attributes, but are required to provide the same drug release characteristics in vivo (humans). In addition, the generic products are also required to provide similar dissolution characteristics, thus forms the basis of pharmacopeial (USP) testing.
Therefore, developing or conducting an in vitro dissolution test just to evaluate/assess differences in formulation/manufacturing, without its link to in vivo, is of limited value or use and may be an incorrect practice.
The purpose of any analytical method development, including dissolution testing, is to have a method that would describe the unknown property of the material it tests. The foremost requirement for method development practices is that one must have reference material, or a product, of the known value of the parameter or dissolution result. A method will be considered developed when this method provides the value of the reference product’s parameter (dissolution results) accurately with acceptable confidence (variance).
Unfortunately, in the case of drug dissolution testing, no such reference product is available with known or accepted dissolution value/result. Thus, in the true sense of the meaning of method development, it is impossible to develop a dissolution method that could be used to determine the unknown dissolution results of a test product.
On the other hand, current practices use the terminology of method development to choose apparatuses and associated experimental conditions that would reflect the expected or desired behavior of the test product. Another way of saying this is, the test product becomes its own “reference.” That is why products often come with their methodologies. Obviously, it is not the correct and valid understanding and method development approach, thus requiring reconsideration. An obvious outcome of such practices is that the developed method would not allow the comparison of drug dissolution (release) characteristics between products. In addition, one would never know the true dissolution rate (characteristics) of any product.
Therefore, in reality, current practices of method development in drug dissolution testing are neither accurate nor serving their intended purpose.
IVIVC (In vitro-in vivo correlation) is a desired feature in the practice of drug dissolution testing. An appropriate IVIVC provides credibility to an in vitro dissolution test by avoiding false-negative indications concerning the quality and manufacturing of a product. In addition, it further enhances economic benefits to the manufactures by providing efficient development and modification of products, thus obtaining regulatory approvals.
Developing an IVIVC may be considered a two-part process: (1) analyzing the in vitro dissolution data and relating it to in vivo results. This has been the subject of the last few posts (1, 2, 3); (2) conducting an actual dissolution test for generating appropriate data. This post is regarding the latter aspect.
For an appropriate dissolution test, in general, and particularly for developing IVIVC, one must conduct the test selecting experimental conditions to simulate an in vivo environment as closely as possible. Commonly the following experimental conditions should be considered in this regard.
A dissolution medium should be an aqueous solution having a pH in the range of 5-7 and be maintained at 37C. The expected amount of the drug present in the product must be able to freely dissolve in the volume of the medium used, often 900 mL. If the drug is not freely soluble in water, then small amounts of a solubilizing agent such as SLS may be used.
The dissolution medium should not be de-aerated. Instead, preference should be given that the medium equilibrated at 37C with dissolved air/gasses, particularly for IVIVC studies.
An apparatus should be selected to have an appropriate mechanism to provide thorough but gentle mixing and stirring for an efficient product/medium interaction. The use of sinkers may be avoided as these often alter the dissolution characteristics of the test products. Paddle and basket apparatuses are known for their inefficient stirring and mixing. Thus their use should be critically evaluated before use for IVIVC studies.
Frequent samples (8-10) should be withdrawn to obtain a smooth dissolution profile leading to complete dissolution within the dosing interval of the test product in humans.
If the dissolution results are not as expected, then the product/formulation should be modified to obtain the product’s desired/expected release characteristics. However, altering experimental conditions such as medium, apparatus, rpm, etc., should be avoided as these are generally linked to GI physiology which remains the same for the test to test or product to product. Therefore, obtaining dissolution results by altering testing (experimental) conditions may void the test for IVIVC purposes.
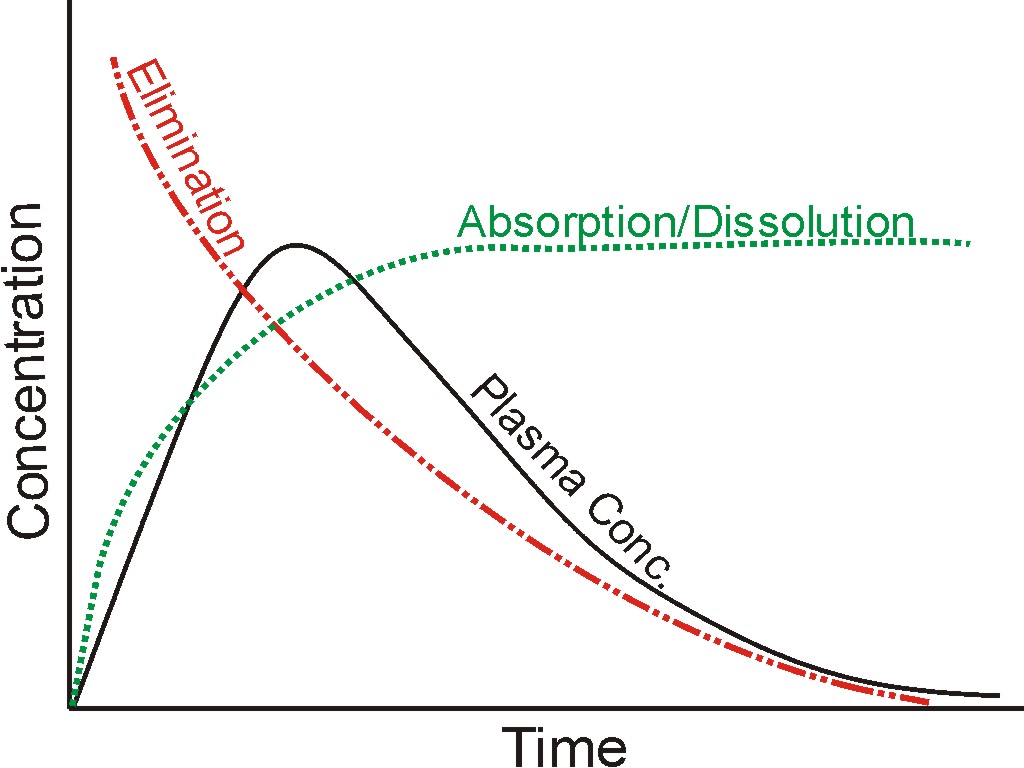
As explained in an earlier post, commonly used convolution/deconvolution techniques for IVIVC purposes link three functions together. The three functions are input (absorption/dissolution results), output (plasma drug concentrations), and weighting function (usually plasma drug concentrations following an intravenous dose).
Deconvolution will be the option one would use when plasma drug concentrations of the test products are available, and one would like to determine the in vivo dissolution results. These in vivo dissolution results are compared with the in vitro dissolution results.
Convolution will be the option one would use when in vitro dissolution results are available, and one would like to determine plasma drug concentrations of the test product. Furthermore, a convolution technique would be the only choice during the product developmental stage, where a formulator likes to have an idea of potential in vivo output. Again, one may use the convolution technique to compare release (dissolution) characteristics of products for generic developments or product modifications. In this case, based on obtained dissolution results from two or more products, one would obtain respective plasma drug concentration profiles, which can be compared using standard and accepted parameters of Cmax and area under the plasma drug concentration curves (AUC).
Studies have shown that the convolution technique provides better accuracy of outcome (plasma drug concentrations) than the deconvolution technique. Moreover, computation-wise, the convolution technique could be simplified, and calculation may be performed using simple spreadsheet software rather than complex mathematical software. Therefore, it is suggested that one may consider convolution as a first choice for developing IVIVC.
A plasma drug concentration-time profile is usually the net effect of two simultaneous processes: (1) absorption of the drug from the GI tract. As absorption is proportional to drug dissolution thus absorption and dissolution are used interchangeably; (2) elimination of the drug from the blood. These two actions, and their net effect, are represented by three profiles and are shown in the figure.
In mathematical terminology, these three curves (profiles) are functions: dissolution or absorption as input, blood concentrations as output, and elimination as the weighting factor or function. To further simplify, in the analogy of linear regression used for calibration curves, output function may be considered as “Y or dependent variable,” “X or input function,” and “M or slope/proportionality constant.” In linear regression analysis, X, Y, and M parameters have values (numbers); however, these are functions in the case of drug concentration profiles. So solving these function-based equations is a bit more complicated.
The procedure is similar to that of linear regression, commonly used to establish calibration curves and then use the calibration curve to determine the unknown concentrations or response (e.g., absorption or peak height/area values). So, if “Y” is known, then “X” may be determined and vice versa. Similarly, if the input function is known, one can determine the output function and vice versa. Determining output function (plasma blood concentrations), if input function (dissolution results) is available, the procedure will be called convolution technique. The inverse of it that is obtaining input function (absorption/dissolution results) if output function is provided, the procedure will be called deconvolution.
There are computer software available that provides the capability of solving for a function when the others are available. However, the convolution approach could be simpler where commonly available spreadsheet software may also be used. For further detail see, Qureshi, SA. In Vitro-In Vivo Correlation (IVIVC) and Determining Drug Concentrations in Blood from Dissolution Testing – A Simple and Practical Approach. The Open Drug Delivery Journal, 2010, 4, 38-47. (Link)
Theoretical Consideration: The most commonly used definition of IVIVC (In Vitro/In Vivo Correlation) is the one described in one of the FDA guidance documents (link). It defines IVIVC as a predictive mathematical model describing the relationship between an in vitro property of a dosage form (usually the rate or extent of drug dissolution or release) and a relevant in vivo response, e.g., plasma drug concentration or amount of drug absorbed.
In this regard, the most sought-after relationship is of “Level A.” It is defined as a predictive mathematical model for the relationship between the entire in vitro dissolution/release time course and the entire in vivo response time course, e.g., the time course of plasma drug concentration or amount of drug absorbed.
Practical Consideration: On the practical side, the purpose of IVIVC is to use drug dissolution results from two or more products to predict similarity or dissimilarity of expected plasma drug concentration (profiles). Before one considers relating in vitro results to in vivo, one must establish similarity or dissimilarity of in vivo response, i.e., plasma drug concentration profiles. The methodology of establishing similarity or dissimilarity of plasma drug concentrations profile is known as bioequivalence testing. There are very well-established guidances and standards available for establishing bioequivalence between drug profiles and products.
Ideally, therefore, one should focus on predicting or calculating plasma drug concentration profiles from drug dissolution results for an appropriate IVIVC. A common mathematical technique employed for this purpose is known as convolution, which convolutes (combines) dissolution results (profile) with plasma concentrations following intravenous (IV) drug administration to provide expected plasma concentrations for solid oral dosage forms. In mathematical terminology, dissolution results become an input function, and plasma concentrations (e.g., from IV) become a weighting factor or function, resulting in an output function representing plasma concentrations for the solid oral product.
Further details about this methodology and its use will be described in future posts.
In continuation of the earlier post (link), this post describes some of the steps commonly described in the literature as method development but should not be considered method development steps. These steps are usually variations in experimental conditions to achieve certain desired characteristics of a dissolution test such as discriminatory, improved reproducibility and/or bio-relevancy. The variations can be are numerous, such as choice of apparatus (paddle or basket), spindle rpm (50, 75, or 100), media (water or buffer), pH (between 1 and 6), choice of de-aeration, or its technique, use and choice of a sinker, etc.
Considering these steps or practices as method development is incorrect because of two reasons:
1. These practices are commonly used during the product development stage. At this stage, an analyst is working with the variations in the product (formulation and manufacturing attributes), which requires a fixed dissolution method, not variations in the method itself, to evaluate the impact of product variations. 2. The products are developed for human use. The drug is expected to be released from the product in the GI tract environment, which remains constant, from product to product. The in vitro dissolution testing conditions simulate this GI tract environment. Therefore, these conditions should also remain constant.
It is, therefore, critical that a dissolution test method should be decided and fixed, reflecting GI tract environment, prior to working on the development of a product.