
For further details on the topic, please see the following links: 1, 2
Commonly described dissolution methods are product-dependent. Therefore, it is not possible to know whether the observed dissolution characteristics are a reflection of the product (formulation and/or manufacturing) attributes or because of the experimental conditions (apparatus, rpm, medium, etc.) employed.
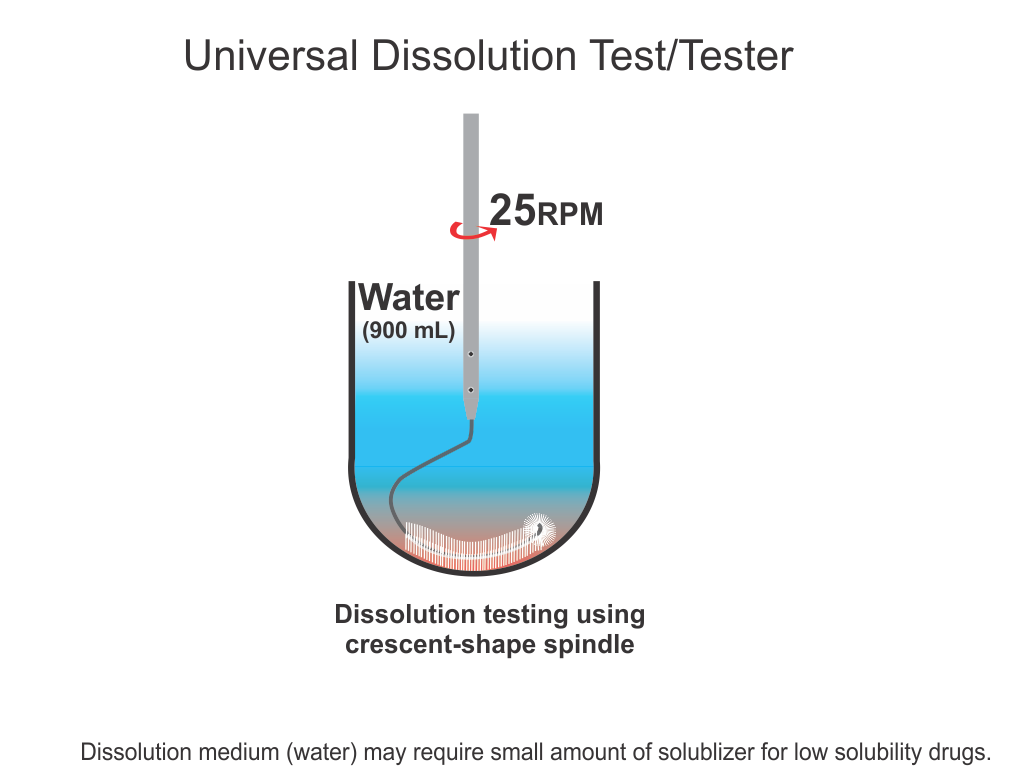
For an appropriate and accurate assessment of dissolution characteristics of a product, the dissolution method must be product-independent. The use of crescent shape spindle has been suggested based on this principle, thus providing true product dissolution characteristics. Please see the following links for further discussion:https://bioanalyticx.com/simpler-and-more-appropriate-dissolution-testing/
(2) http://www.drug-dissolution-testing.com/blog/files/Flyer.pdf
(3) https://bioanalyticx.com/product-dependent-dissolution-testing-a-scientifically-invalid-practice/
(4)
The objective of drug dissolution testing is to determine drug release characteristics of a product i.e. how fast or slow a drug would be released from the product reflecting its formulation and/or manufacturing attributes (properties).
In reality, however, current practices of dissolution testing seek product-dependent experimental conditions based on choices of apparatus, rpm/flow rate, and medium (its pH and strength) to attain a desired or expected dissolution rate. “True” dissolution characteristics of a product can never be known. Using the current dissolution practice or approach, multiple dissolution methods are often described for the same product under different names such as bio-relevant method, discriminating method, QC method, USP method, FDA method, etc. differing in experimental conditions, each providing different dissolution results.
That is why it is commonly referred that one should consult the authorities to establish the method and to determine if the results would be acceptable to the authorities.
For establishing “true” dissolution characteristics, like any other analytical method, a dissolution method must be product-independent. Developing a product independent test leading to determining “true” dissolution characteristics of a product is relatively simple, saving significant financial and human resources compared to the current practices. For further discussion on this aspect, please see the article and details of the upcoming course.
Drug dissolution test as a quality control test: In simple terms, at present, a dissolution test as a QC test means conducting a dissolution test as described in a pharmacopeia, in particular USP. If the test meets the pharmacopeial (Tolerances) requirements, the product may be considered a “Quality” product. It is, however, not clear what “quality” the test refers to or to what product property the test is linked to? Therefore, to overcome this lack of objectivity/relevancy, pharmacopeial tests are described as “consistency” tests. Again, it is not clear the “consistency” of which property or parameter the test is referring to?
Dissolution testing during product development: One of the main uses of dissolution testing is to facilitate product development. This use is based on the principle that the tests should be able to provide potential in vivo drug release behavior information. However, this is a commonly recognized fact that currently used dissolution tests generally do not provide in vivo relevant product characteristics. Thus, current practices of dissolution testing at the product development stage appear to lack relevancy and objectivity.
Practices of methods development: Developing a method requires a well-established and accepted reference (product or parameter). In this case, a reference product should be available with known drug dissolution results established independently. As there is no reference product with known dissolution results available at present, it is not possible to develop a dissolution method. As commonly suggested in the literature, developing a method based on test products (with unknown dissolution characteristics) is neither scientifically valid nor possible.
Dissolution apparatus selection: Selecting adissolution apparatus should also be part of a method development exercise. An apparatus must be able to provide a relevant testing environment. Current practices appear to suggest that relevant apparatuses, and associated experimental conditions, are those which are described in the pharmacopeias only. On the other hand, it has never been shown that suggested apparatuses and the associated experimental conditions are suitable for their intended purpose. That is, the suggested apparatuses have never been validated for the intended purpose.
Selecting experimental conditions: Choices of experimental conditions such as rpm and buffers described in the literature appear to be arbitrary and random. Moreover, experimental conditions are selected to achieve some desired dissolution characteristics of the test products. Thus, a formulator/analyst would never know the “true” dissolution characteristics of the product. Experimental conditions must be linked to the physiological environment and be product-independent. Drug and/or product-dependent experimental conditions, as commonly described, are neither scientifically valid nor relevant.
Establishing IVIVC: Developing IVIVC commonly refers to RELATING in vitro dissolution results to in vivo dissolution results using a mathematical approach of de-convolution.However, in general, dissolution tests are conducted based on the principle that in vitro release should relate well with in vivo results. Therefore, it is unclear why every manufacturer/analyst has to go through this exercise of establishing this in vitro-in vivo link for each product. In addition, as stated above, the current apparatuses/practices of dissolution testing have never been validated for providing relevant in vivo dissolution characteristics, so why should one be expected to achieve appropriate IVIVC?