
Further details: (link), (link), (link)


Recently (July 21, 2021), CDC announced that it intends to withdraw Emergency Use Authorization (EUA) of the CDC 2019-Novel Coronavirus (2019-nCoV) Real-Time RT-PCR Diagnostic Panel, or in simple language, withdrawing its recommended PCR test (Continue here)


For details, please see here Link

People, including mainstream “scientists” and “experts,” do not realize that vaccines, mRNA-based or otherwise, have never been tested for their efficacy. To test the efficacy for developing and testing the vaccines, the virus (SARS-CoV-2) must be available in pure isolated form. This is not only a scientific requirement but simple logical consideration as well. It is impossible to establish the usefulness and effectiveness of the vaccines without the use or presence of the target, i.e., the virus.
It is a commonly known fact now that no purified isolated specimen of the virus is available anywhere in the world. Therefore, no one can test the efficacy of the vaccines, and it has not been done either. Saying it otherwise is simply a lie.
The development of vaccines is based on testing against the PCR test, which is not a test for the virus but an RNA/DNA-based marker of the unknown or imaginary virus (commonly known as SARS-CoV-2). The PCR test is an arbitrary “dipstick” type test without any link to the virus, infection, or illness. As the virus has never been isolated, it is impossible to link the marker to it and validate the PCR test for its relevancy and accuracy as well.
Testing and assessing viruses and their link to illnesses and the treatments, as currently described and promoted, reflect ignorance and incompetency of the “experts” and “scientists.” Therefore, the medical and pharmaceutical areas require urgent scrutiny and audit of their scientific claims.
The focus should be treating the illness/infection, if and when it occurs, and not developing the treatments (such as vaccines) for the imaginary virus and its mutants.
For further reading:
COVID-19: The virus does not exist – it is confirmed! (link)
The science behind COVID and vaccines! (link)
COVID-19: Vaccine ‘Not Possible’ For A Virus Not Yet Quantifiable (link)
It appears that the current media indulgence with finding virus origin is not related to the public health policy or benefit but to divert the attention from the fake story of the virus existence. The news or debate about the virus’s origin is indirectly establishing the view that the virus exists. However, science provides no evidence of the existence or presence of the virus.
No scientist or “medical expert” worldwide has provided any valid evidence supporting the existence of the virus. It is a well-known fact that the virus (a ball with spikes) is an imaginary and computer-generated artifact, not physically found in a specific geographical area or laboratory. Therefore how could its origin be determined – it cannot be. The media discussion legitimizes the virus’s presence so that its fear in public should continue and the recommended “treatment” could be promoted.
A more appropriate focus of the discussion or probe should be that if the virus never existed, what have “medical/health scientists” doing for the past so many years with all their “work” and money/grants they received for the virology work?
The disastrous social and economic outcomes of health authorities’ reaction in controlling the pandemic and projected deaths are well known now. The spread and campaign of inciting fear in public have been intense and continuous – all in the name of achieving public health and safety. Authorities continue with their draconian measures, with the advice from medical professionals and experts laced with slogans of following scientific principles and practices.
On the other hand, the current situation raises questions about the legitimacy and correctness of medical professionals’ claims concerning science.
This article provides an argument that medical professionals never gained education and training to conduct scientific research and experimentation. The lack of expertise in science appears to have resulted in false claims about the virus’s existence, isolation, spread and illness, and the development of treatment such as vaccines. It is suggested that in the future, such an issue should be handled by academically trained scientists in relevant fields such as chemistry, especially analytical chemistry (Continue here).
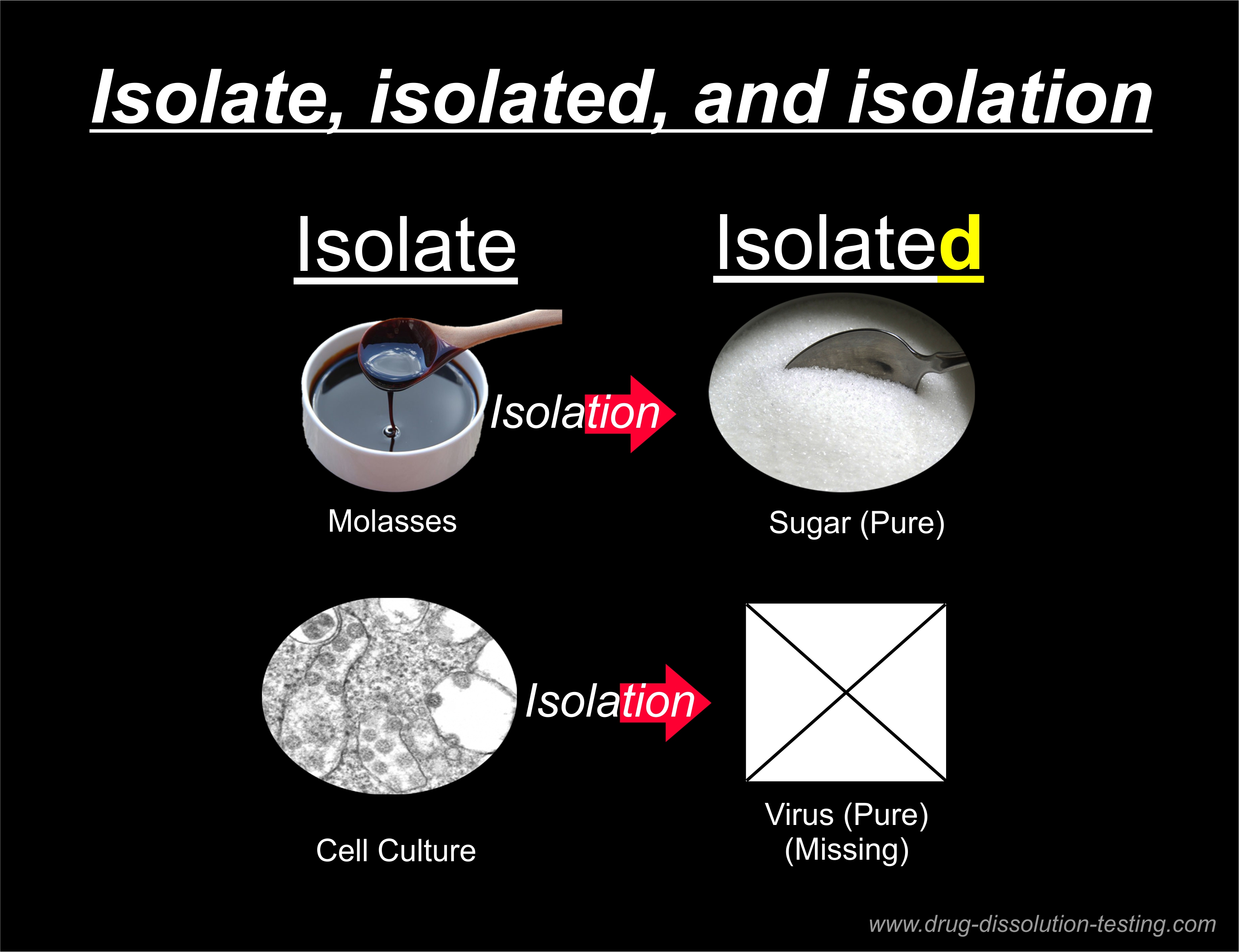
What is needed is an “isolated virus,” which is not available. Every claim without it (virus, PCR test, COVID, pandemic, vaccines, etc.) becomes irrelevant and false.
The pure isolated virus box is empty because the virus is neither available nor exists.
Added note:
The purpose is to clarify the confusion in the virology literature.
The virology literature uses the word “virus isolate” to represent the “isolated virus” as if chicken soup means “chicken,” which is not correct. The “virus isolate” represents (mixture, soup, gunk) while “isolated virus” means a “pure component.”
The process of separating a pure component from the soup or gunk is called “isolation” or extraction. As the pure “isolated virus” is not available, the virus’s isolation step has never been done.
I hope this clarifies the picture and the message it is trying to convey.

