
The virus (SARS-CoV-2) has never been isolated, positively identified, or physically characterized. COVID-19 does not have defined, specific or measurable symptoms and has never been linked to SARS-CoV-2 experimentally (as it has not been isolated and identified). A strong possibility exists that COVID-19 could be an incorrectly diagnosed disease – may be an imaginary one. Therefore, scientifically the implementation of vaccination should be considered an incorrect or false treatment.
Author: Dr. Saeed Qureshi, Ph.D.
As a part of a discussion on LinkedIn (link), I posted a response highlighting grievous misunderstanding and misinformation concerning the PCR test’s relevancy and scientific validity. My posted commentary appears missing from the forum, along with the concurrent disappearance of Dr. Daniel Goldstein’s numerous comments. I believe my analysis is valid and would be helpful to others for future discussions on the topic. The visitors of this blog may also find my post (below) useful as well.
Response to: Daniel Goldstein, MD (Professor & Vice Chairman, Department of Cardiothoracic Surgery Montefiore Health System), link
Please be nice and civil. If you do not respect yourself, consider respecting your profession and the organization you may be representing.
Thanks for pointing out the typo on my blog. I will go through it and will correct it if required. You may also suggest where you found the error specifically, which would be helpful. On the other hand, under discussion is the validation of the PCR test, which is not affected if my blog has some minor typos.
Regarding the post “blocked from LinkedIn” on my blog, I do not consider it a bad thing but a good. Otherwise, I would not have put it there. As I have pointed out before, my views are not in line with current popular belief but are based on independent thinking, expertise, and analytical science experience. This aspect is missing from the present projected and promoted opinions.
Eléna Diakaki is following my work for some time. Based on my description of the issues (virus, testing, illness, pandemic, etc.), as I usually describe in a simple and mostly non-technical language, she, like many others, agrees with me, at least understands the issue well, and are questioning the popular view. If you do not agree with my perspective, or have a different opinion, share your thoughts with supporting science and data in a simple language so that people can understand. There is no need for jumping up and down and shouting. You do not realize that your responses clearly show that you do not have anything valid in support of your argument, except telling just follow others like you are following.
Everyone is working to improve the health and happiness of people. You are not the sole custodian of this aspect. From my perspective, as it is a well-known and well-established fact that PCR tests are flawed and bogus (you may believe or say whatever you like), so the testing must be stopped immediately. The pandemic will disappear in a blink of an eye as well. People are getting labeled (diagnosed) incorrectly with this flawed test. Once the testing stops, patients come to the hospital, assess them accordingly and appropriately, having an open mind.
I hope you will consider the issue under discussion calmly and explain why you believe the PCR test a valid test. BTW, as I mentioned before, you might have difficulty in understanding the question, so please seek some help from someone knowledgeable in the testing area and validation of tests.
In the end, I would say that I may get blocked from LI again. People are and will be doing badmouthing. If that happens, please reach me through my blog (www.drug-dissolution-testing.com). I will be happy to provide my opinion to you or anyone else to the best of my abilities.
Good luck
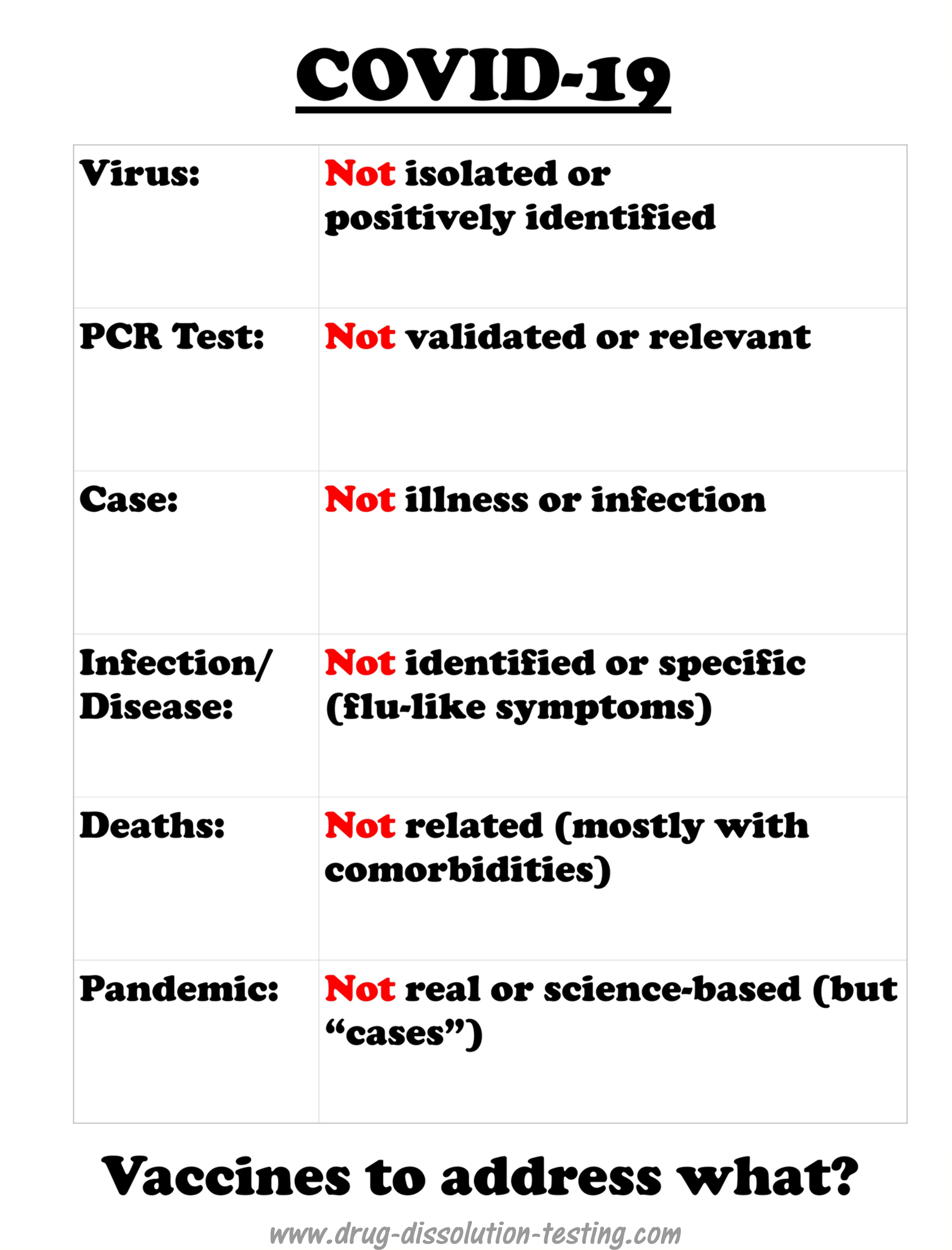
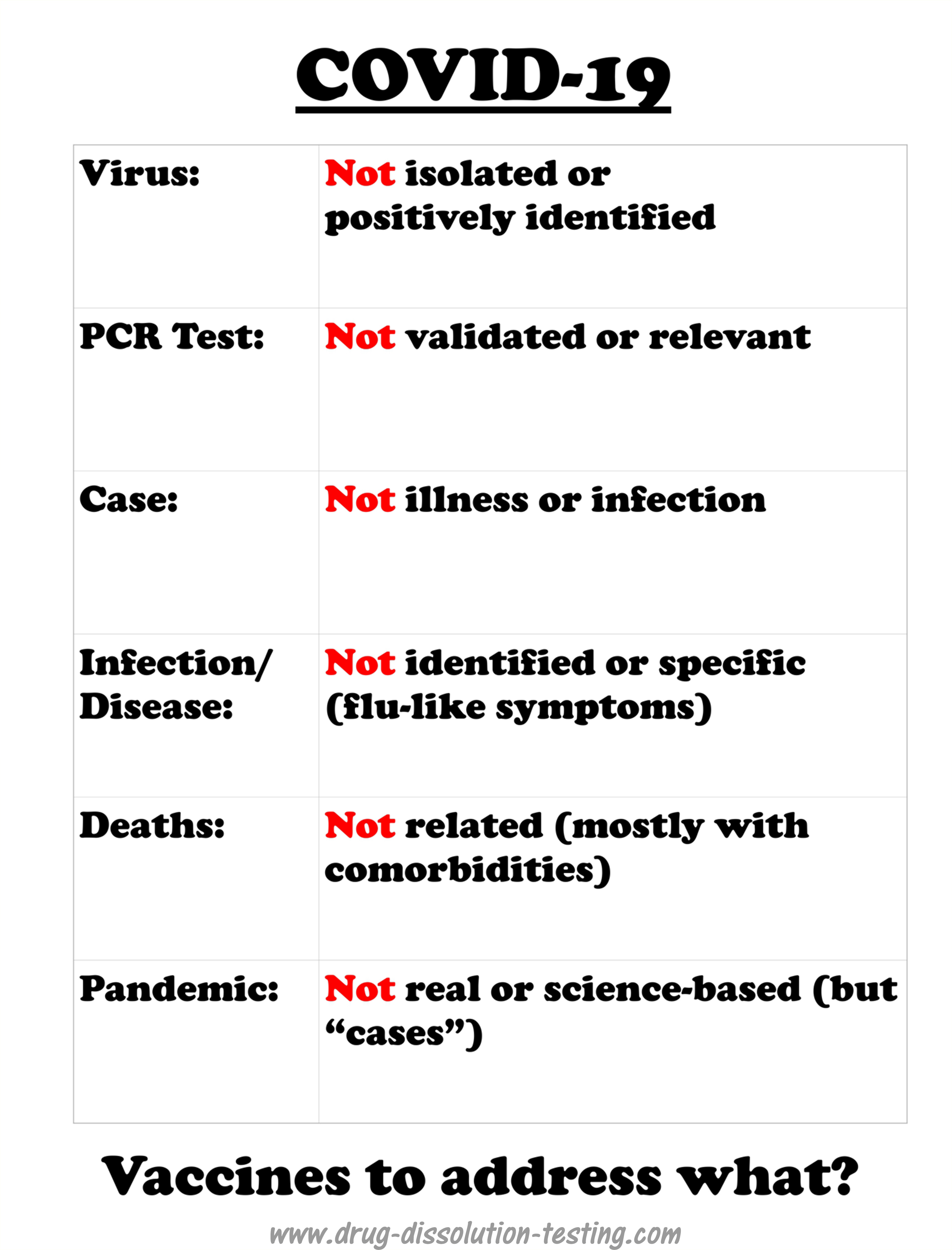

COVID-19 is a recently labeled infectious disease that is presumably caused by a novel coronavirus labeled as SARS-CoV-2.
It is important to note that COVID-19 is not based on any defined and specific symptoms but common and general flu-like potentially treatable with antibiotic regimens [1]. However, medical experts and regulatory authorities, in particular FDA, have adopted an official position that illness is because of a viral infection caused by SARS-CoV-2. Being a viral disease led to a policy decision that a vaccine is needed for its treatment that is to be developed. The pharmaceutical industry has made great efforts in collaboration with the authorities to develop vaccines quickly. There have been media reports that some vaccines are at a late development stage and ready to be submitted to the FDA for marketing approval.
Continue here
One thing is for sure that the test does not show the presence of the virus. Anyone who says otherwise is misinformed or lying. At present, there is no test available to test the coronavirus.
The test, which is usually conducted using swab samples, is called the PCR test. The PCR test does not test the virus, but a long chain chemical compound commonly known by its fancy name DNA or RNA. Therefore, the test is a chemical test to monitor a chemical compound (DNA or RNA) or its part by its nature. With a positive test result, it is assumed that the RNA is from the coronavirus. However, because no one has seen or isolated actual coronavirus, it is impossible to establish if the RNA (or its part) is from the virus. The positive test results could be from any or many sources, including regular seasonal, live or dead, flu-virus, or its debris.
Also, scientifically speaking, before any test is accepted for its intended use, it must be validated to show that it is specific and accurate. However, the current PCR test could not be validated because it would require coronavirus itself and its RNA that is not available. Therefore, the PCR test becomes invalid or irrelevant and unpredictable.
So to the question, “What does a positive coronavirus test really mean?” The answer is NOTHING! For a more detailed discussion on the topic, please follow the link..
(Revised for editorial changes on November 23, 2020).

A few days ago I provided critical comments on a publication from Australia (University of Melbourne) which claimed isolation and identification of SARS-CoV-2 virus [1]. I suggested that claims were not supported by scientific evidence and logic.
The present article critically evaluates a similar claim from America’s Center for Disease Control (CDC) in a publication [2] entitled: Continue here

It is both logical and common sense to expect that if a particular material is claimed to exist then its presence must be established using valid and well-recognized practices and laws of science. For example, if it is suggested that a certain geographical area may provide a significant amount of minerals such as gold or oil then that mineral must be extracted, isolated, and characterized before proceeding with large-scale production for public benefit and commercial gains. The same understanding has to be applied in other areas including the medical and pharmaceutical areas.
At present, the world is allegedly in the grip of a serious and widespread disease (pandemic) referred to as COVID-19 caused by a virus labeled as SARS-CoV-2. Hence, there is purportedly an urgent need for a treatment for this disease. Commensurately, it is important to note that the medical community has declared with apparent certainty that the disease (COVID-19) exists and is caused by the virus SARS-CoV-2. [click here to continue]
Edited and revised for clarity and grammatical improvements on November 5th, 2020.


If scientists and health care professionals, including physicians and pharmacists, at the regulatory authorities had paid attention to false testing practices and requirements for pharmaceuticals, current pandemic would never have happened. Basis of virus and pandemic is “testing” developed and implemented by experts with no or limited expertise in the area of testing resulted in false conclusions and declaration of the virus just like persistence false claims about the quality (by extension safety and efficacy) of the approved and marketed pharmaceuticals (1, 2).
It is impossible to get out this situation until analytical science (testing) and its principles are allowed to be followed. Fearmongering and censorship have always been tried like at present to protect the status quo and the “experts” however they always fail – so will be this time as well.
Consider conducting testing based on relevant and true scientific principles.