
The unfortunate situation created by this Coronavirus pandemic is providing a serious opportunity for reassessing the current regulatory approaches in pharmaceutical products development and their manufacturing so that such irrelevant discussion can be avoided and patients can have access to modern and multiple options to treat ailments. Hopefully, in the future, patients will be treated with well-established products rather than products developed on the fly or with the use of disposable gowns, masks, washing hands and/or staying home policy which certainly is not the treatment. Patients expect and deserve something better from us as scientists, physicians, and regulators. Follow the link for the complete article (link)
Author: Dr. Saeed Qureshi, Ph.D.
Yesterday, I posted my view on the recent FDA guidance documents for chloroquine and hydroxychloroquine (link). I do not think people realize the long-term impact of this development where BE studies have been replaced/substituted with drug dissolution testing. Let me explain:
- Saying that guidances are product specific is not correct because chloroquine and hydroxychloroquine are drugs not products. Products are tablets, capsules, often unknown and proprietary compositions of a drug, excipients, and manufacturing attributes (i.e. formulation and manufacturing attributes). Hence, guidances cannot be product-specific as assumed or suggested.
- A drug dissolution test is conducted for products, not drugs. As product attributes are mostly unknown and propriety, as noted above, hence a dissolution test (or guidance) cannot be product specific but has to be independent “standard or universal.”
- Furthermore, it is to be noted that the product-specific guidance concept is an invalid concept in principle. Drug dissolution testing is a scale used to measure the dissolution characteristics of a product. By definition, it (scale) has to be independent of the tested items. The point being that the guidance documents cannot be restricted to one or two drug products. These have to apply to ALL highly soluble drug products. It would not be possible for authorities, at least scientifically, to defend restricting to only one or two products. This decision could easily be challenged and won.
- In addition, such a decision cannot be a one-time decision, as many believe, may it be taken under an emergency. It would not be possible to withdraw such a decision once taken i.e., if dissolution test alone can provide a quality assessment of the products, then why would BE studies be needed and required on what basis, especially when BE studies are known to be irrelevant (link).
- This new development is a gift from heaven for the underdeveloped countries where because of lack of BE studies, products and their manufacturing have always been labeled inferior. However, with the dissolution testing only, manufacturers can manufacture and promote (for local and/or international markets) their quality products with confidence.
Keep these thoughts in mind and proceed accordingly.
Recently FDA provided 1- and 2-pager guidance documents for chloroquine and hydroxychloroquine, respectively (link).
The most interesting part is that one can get product approval based on dissolution testing alone. This is what has recently been suggested in one of my recent published articles, i.e., products (“quality”) assessment can easily and accurately be established with drug dissolution testing alone (link). Therefore, there is really no need to conduct bioequivalence (BE) assessments. These (BE) assessment procedures have never been validated of the intended purpose. In fact, BE is scientifically invalid and can provide false conclusions and assurance about product quality. In addition, such testing exposes healthy human volunteers to highly potent chemicals under the disguise of medicine development.
On the other hand, switching to dissolution testing alone using currently recommended USP apparatuses is not valid either, at least scientifically. The recommended apparatuses are non-GMP compliant and can provide false and irrelevant results because of their intrinsic design and operation problems. Simpler and scientifically valid options are available and could be used (link).
Mortality in the United States, 2018 (as of January 2020, link).
“The age-adjusted death rate decreased by 1.1% from 731.9 deaths per 100,000 standard population in 2017 to 723.6 in 2018.” i.e., death rate is about 0.7236%
For the USA, having a population of 331 million (link), the normal/standard death (attrition) rate should be 199,593 deaths/month. Now compare this number with the reported number of deaths caused by the Coronavirus pandemic, which is 21,435 in about a month as of April 12, 2020 (link), which is far less than the normal/standard death (attrition) rate.
The death rate, therefore, does not appear to support the thesis that the pandemic is killing people in abnormally high numbers.
This morning I received the following query (my response is included below) about the manufacturing aspect of the above-mentioned drug product. It is hoped that authorities will take note and address the issue faced by the industry to manufacture this essential pharmaceutical product as per my year-and-a-half-old Citizen Petition (link).
Query:
“Regarding the dissolution of HYDROXYCHLOROQUINE SULPHATE TABLETS, the disso medium specified in IP & USP is water. The formulation sometimes fail to conform to IP and even USP parameters
I have tried replacing water with 0.1 M hydrochloric acid as dissolution medium and achieved a disso of above 90 %. I know the api is water soluble, but since this is an instant release formulation and the approximate pH of stomach is being maintained in disso medium, can we recommend change of disso medium from water to 0.1 M H Cl to IP & USP.
Response:
Scientifically speaking, water is a suitable dissolution medium, not the HCl (link).
In reality, your suggestion of changing the medium from water to HCl is for obtaining desired dissolution results, which is neither scientific nor logical.
In general, the issue you are describing is not of medium choice but the choice of the dissolution apparatus. USP apparatuses are known to provide slower and irrelevant results; most likely, you are observing this flaw. Such an issue can only be addressed by changing the tester, not the medium, rpm, etc. Considering USP apparatuses’ flaws and limitations, I have proposed a new stirrer to address this issue. Perhaps consider using this suggested spindle and simpler dissolution method not only for this product but also to avoid future issues with USP apparatuses. Additionally, there is a stronger argument for using an alternate dissolution tester than changing the dissolution medium, i.e., the USP apparatuses are non-GMP compliant (link, link).
I hope you will find the suggestion useful, and best of luck.
PS: Please request the authorities, in particular FDA and USP, to withdraw the requirement of using non-GMP (i.e., non-validated/non-qualified testers/methods) and allow the use of scientifically valid testers and methods to develop and manufacture urgently needed pharmaceutical products.
… provides in-depth discussion on establishing and monitoring the quality of pharmaceutical products in particular tablets and capsules. I hope you will find the chapter useful.
Chapter title: “Quality” of pharmaceutical products for human use – underlying concepts and required practices, published in
Drug Delivery Trends: Expectations And Realities Of Multifunctional Drug Delivery Systems Volume 3 Edited by Ranjita Shegokar, PhD., Available from Amazon (March 18, 2020), link.
Reasons:
(1) They do not define the quality of the products. Hence it cannot be measured and/or established (link).
(2) Suggested methods and procedures lack scientific relevancy and validity (link)
(3) GMP practices, including inspections, are about the operation of manufacturing, not per se reflection of product quality (link).
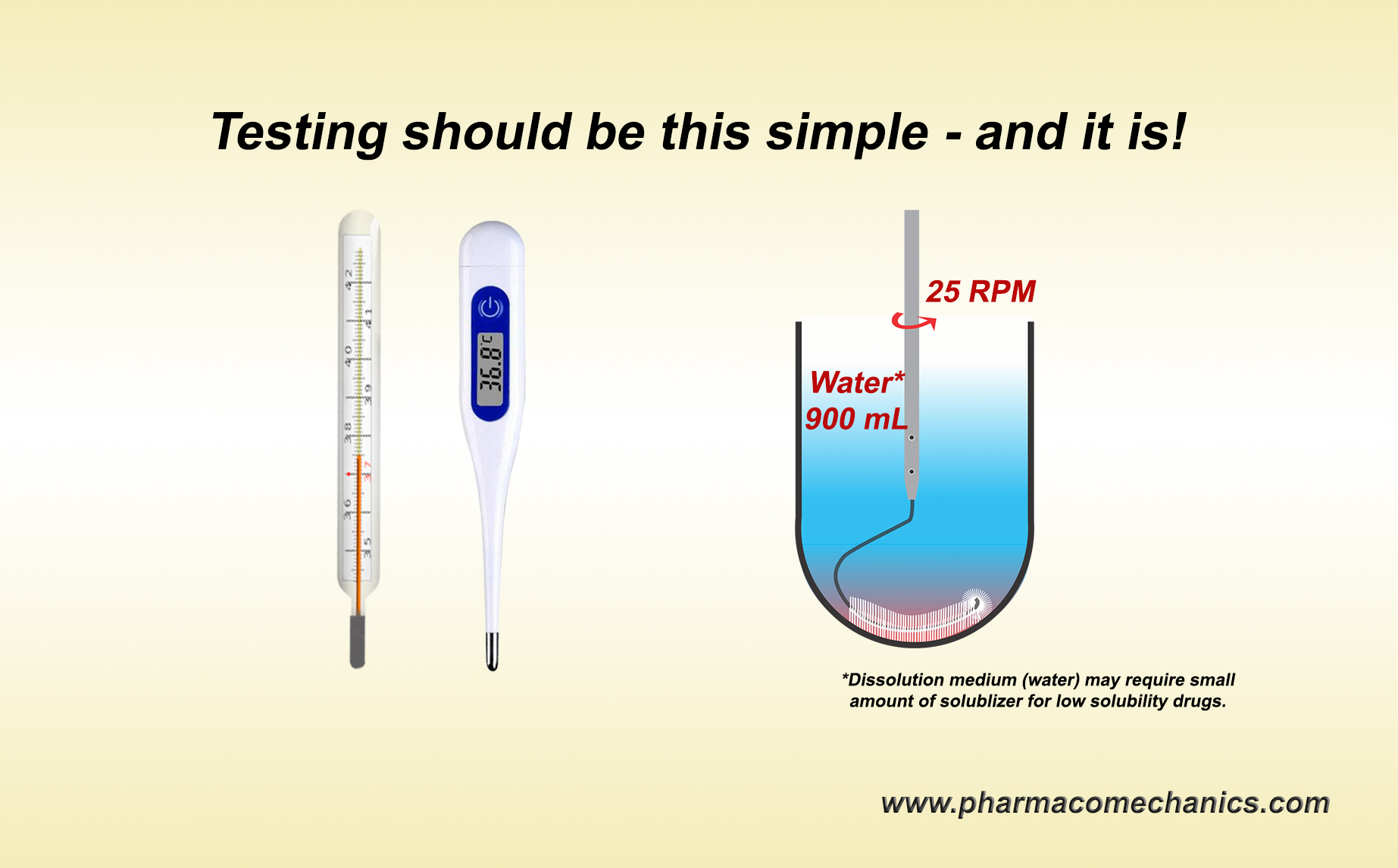
A discriminatory test, in general, is a test or procedure which would measure a given parameter and would be able to differentiate/discriminate between tested objects where the differences exist. A common and simple example of such a procedure or routine is monitoring body temperature using a thermometer. A thermometer is usually placed in the mouth, and after a bit of waiting, the thermometer provides a body temperature reading. Thermometers are pre-standardized/calibrated against reference standards (temperatures). There is no thermometer or monitoring procedure developments, no discriminatory test/procedure development, no “life-cycle” variations in procedures, and no subject-dependent (gender, age, race, body weight/type) procedures. Note that a thermometer does not even tell if the person has a fever. It will only tell body temperature, which is interpreted as whether the person has a fever. Point being a pre-standardized/calibrated thermometer by default becomes a discriminatory test or tester. This is how life/science works.
Similarly, a pre-standardized/calibrated dissolution test/tester must provide a product’s drug dissolution/release characteristic (tablet/capsule). Placing a product into a vessel equipped with a stirrer with pre-set rpm containing pre-defined solvent, its volume, and temperature is the dissolution test/tester. Determining drug release after a pre-defined time would provide the answer, i.e., drug dissolution characteristics of the product. Period! There should be no need for dissolution methods development of any type at the product/formulation development and/or evaluation stages.
Why do we have or need dissolution method development practices, including discriminatory methods – pure ignorance and incompetency of the subject? This is simply nonsense and fake-science! Be clear that no one is currently determining the dissolution characteristics of any product, hence the quality for which this test is conducted and promoted.
Seek sensible and scientifically valid solutions! (1, 2, 3, 4)

The quality of a drug product, such as a tablet and capsule, may be defined as the product’s ability to release the drug in humans in the expected amount and consistently. In technical terms, it is known as the drug release characteristics of the product. This characteristic is established at the manufacturing stage by conducting an in vitro test known as a drug dissolution/release test. The test is commonly conducted as recommended by regulatory agencies (such as FDA) and pharmacopeias (such as USP) using paddle and basket apparatuses. The quality assessment of most of the solid dosage forms, if not all, in particular, tablet and capsule products, is determined using this test.
The testers for this test, however, have never been validated for the intended purpose. Therefore, conclusions drawn from this test, hence the quality of the products, would be invalid and false. Use caution in accepting and/or making claims about the quality of products! (link)
Seek help for conducting a scientifically valid drug dissolution test and establishing the quality of the drug products.
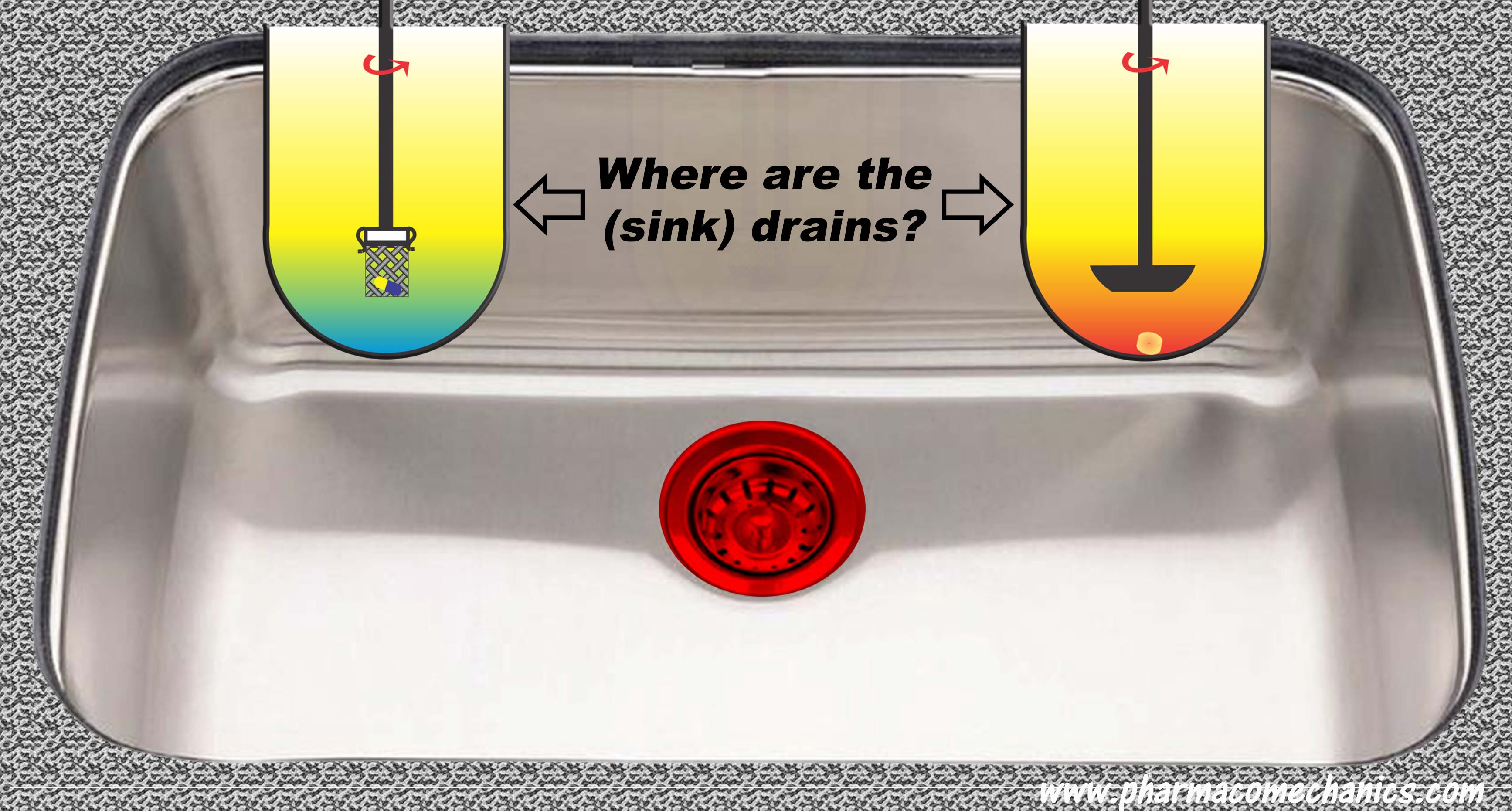
Drug dissolution testers, particularly the most commonly recommended ones (paddle and basket), are based on a closed system/environment, i.e., without a drain. Therefore, it is not possible to have or create a sink condition. Yet, authorities and pharmacopeias require it while “experts” create it. How? Magic!
In addition, one does not require a sink condition for in vitro drug dissolution testing. 900 ml of water with or without a small amount of solubilizer sufficient to (freely) dissolve the expected amount of drug present in the product – is all one needs. For further details, please follow the link to simplify your life and avoid wasting time and resources (link).