In my view, the collapse of the regulatory pharmaceutical science as we know it is coming because it is reaching the limits of promoting fake science, regulatory bullying, and disregarding consumer/patient needs and affordability.
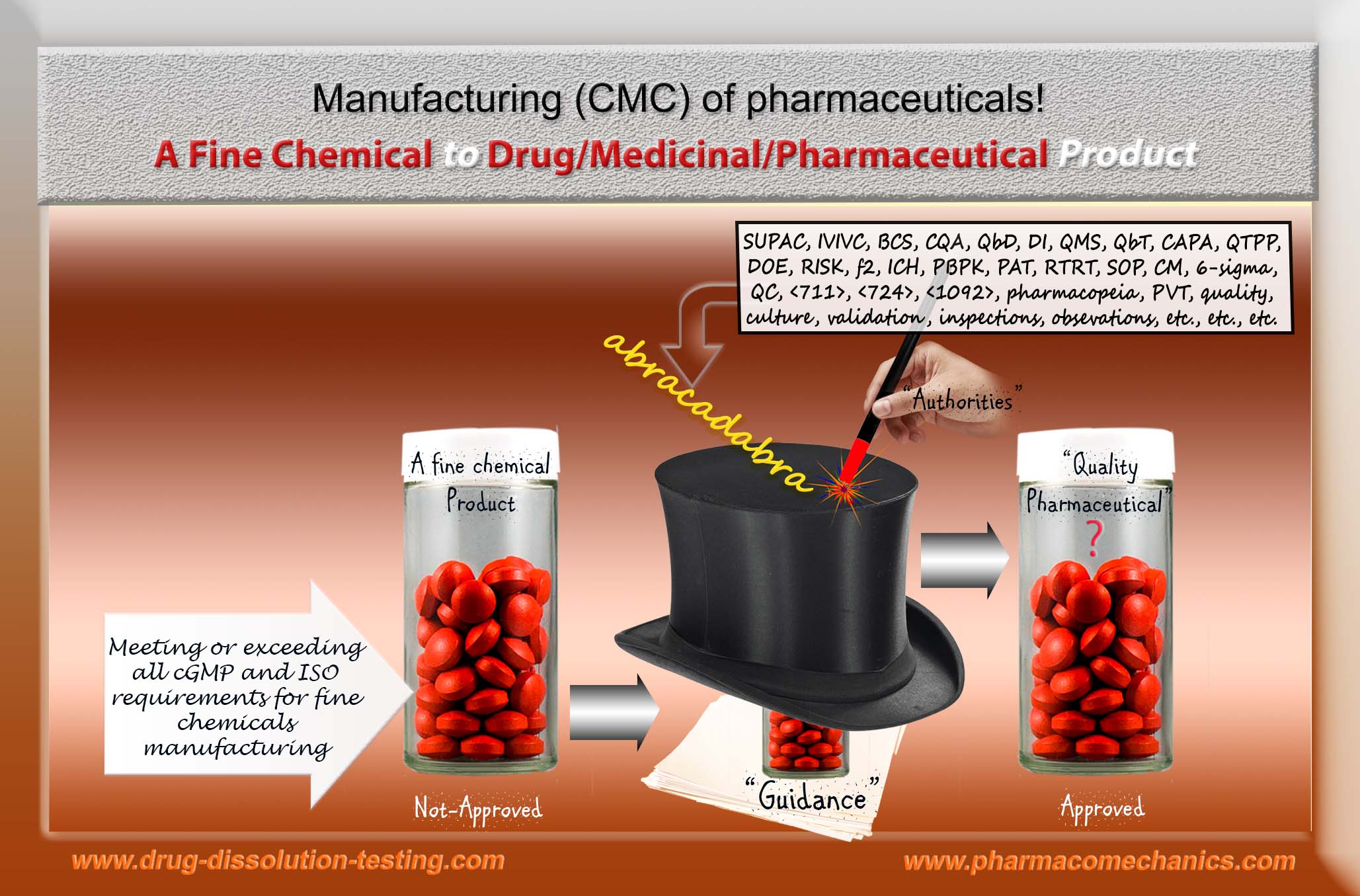
To begin with, it is not even possible to know what one means by regulatory pharmaceutical science or regulatory science. It may be considered a hodgepodge collection of documents and checklists, often termed Regulatory Guidance/Guidelines or pharmacopeial monographs, which like-minded groups of people compile them. The main promoted objective of such exercises is to provide “care or help” to customers/patients by establishing the “quality” of pharmaceutical products, such as tablets/capsules, which should be available at reasonable prices. The irony is that no definition or criterion is provided for evaluating the quality of products. The regulatory authorities and pharmaceutical community never defined what a quality pharmaceutical product is. Compliance with guidance reflects quality without an established link between the two (compliance and quality). Thus, everything related becomes illusionary and scientifically invalid, requiring extraordinary efforts, mostly bullying, to promote and maintain the pretense of patients’ safety and product quality
There is, thus, an urgent need for reassessing current regulatory practices so that the manufacturing of pharmaceutical products and their assessments start to make sense and consumers/patients have access to affordable quality pharmaceutical products. Some ideas and suggestions, as links to some articles, are provided in this regard.
Issues:
(1) Defining roles and practices of pharmaceutical regulatory authorities (Link)
(2) In-compliance with regulatory standards and requirements do not necessarily mean that a pharmaceutical product (tablet/capsule) or process is of quality! (Link )
(3) In all seriousness – this is really an “abracadabra” exercise! (Link).
(4) Quality assessment and prevailing illusions! (Link)
(5) KABOOM! Pharmaceutical Product Quality Issue (Link)
(6) Something to think about! (Link).
(7) Bio-waivers! (Link)
(8) Inspections and quality of pharmaceutical products and/or manufacturing processes – dilemma! (Link)
(9) Fashionable Nonsense (Link)
(10)Non-GMP compliant dissolution testers but no warning letters! (Link)
(11)A Quality Product – Please Define! (Link).
(12)Let me be bold and direct! (Link)
(13)Assessing quality of pharmaceutical products! (Link).
(14)Scientific and GMP violation! (Link)
(15) Guidance Documents – Deficiency (Link )
(16)f2 – Similarity Factor (Link)
(17)If one likes that a manufactured drug product should be of quality, then one needs to define (tell) what a quality product is? (Link).
Suggested Solutions:
(1) Quality of Pharmaceutical Products (Link)
(2) The missing quality definition or metric (Link)
(3) Product Quality Metric (do not confuse it with drug/medicine quality) (Link
(4) Universal Dissolution Test/Tester (Link)
(5) Universal Dissolution Test/Tester – Second Part (Link)
(6) Promoting quality standards for drug products: Scientifically speaking, please be systematic and logical! (Link)
(7) Establishing safety, efficacy and quality of drugs and drug-products (tablet/capsule) – serious confusion! (Link)