

For details, please see here
For details, please see here
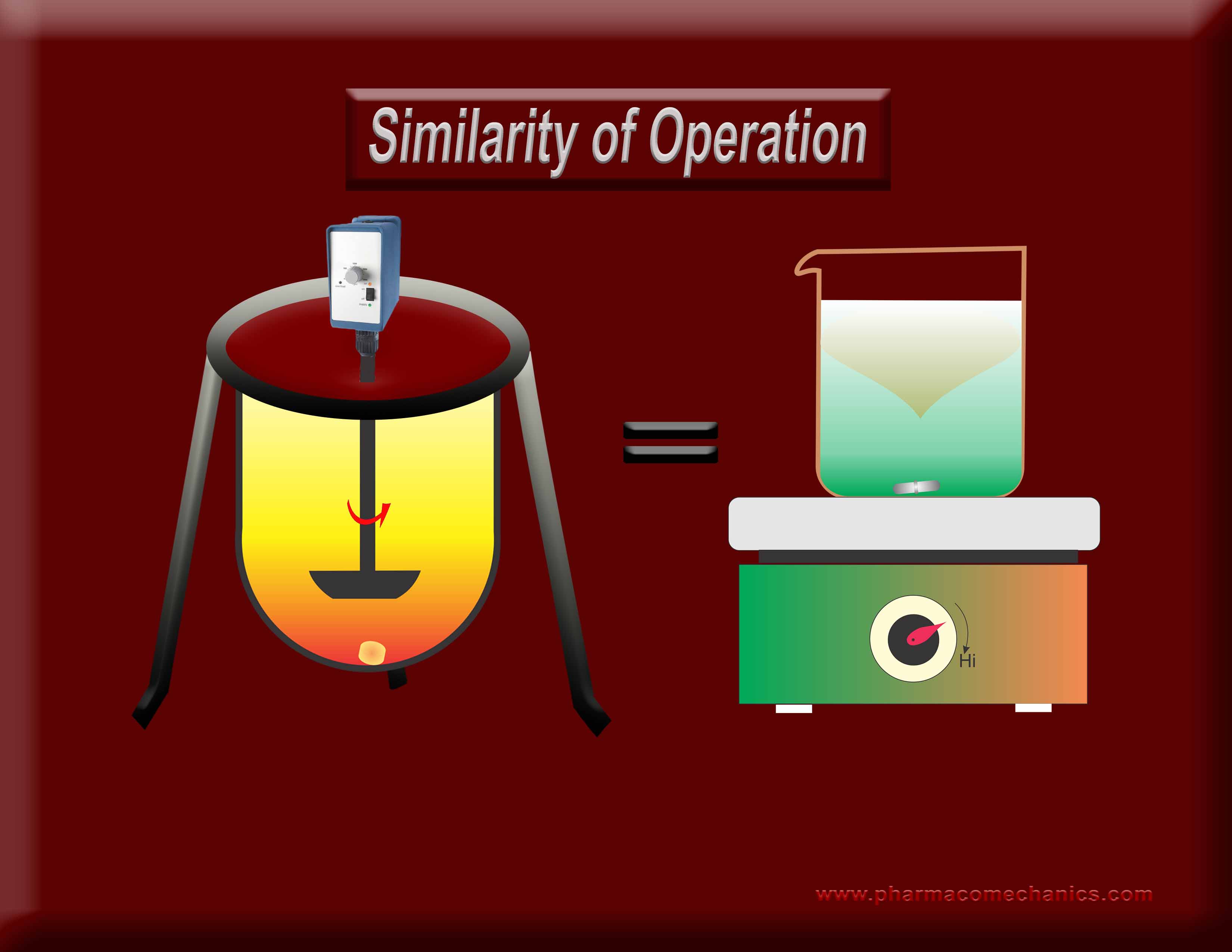
Why does one require an exhaustive standardization (calibration/PVT/Guidances/training etc.) for the operation of one stirrer type but not for the other when both are used extensively for the same purpose (dissolution)? Does it indicate a misunderstanding of the underlying concept? It appears so! To know more about why both of these stirrers are not suitable for DRUG dissolution testing? See here

This definition/metric needs to be included, as an assessment of practically all other factors such as safety, efficacy, manufacturing and its design and processes etc., are secondary to it. In addition, such a metric can easily be explained to the consumer/patient. For further discussions on the topic, please see the link.
Attached is a copy of a presentation (slides) given last month at Rowan University (New Jersey, United States) as a guest speaker for an online course (link).
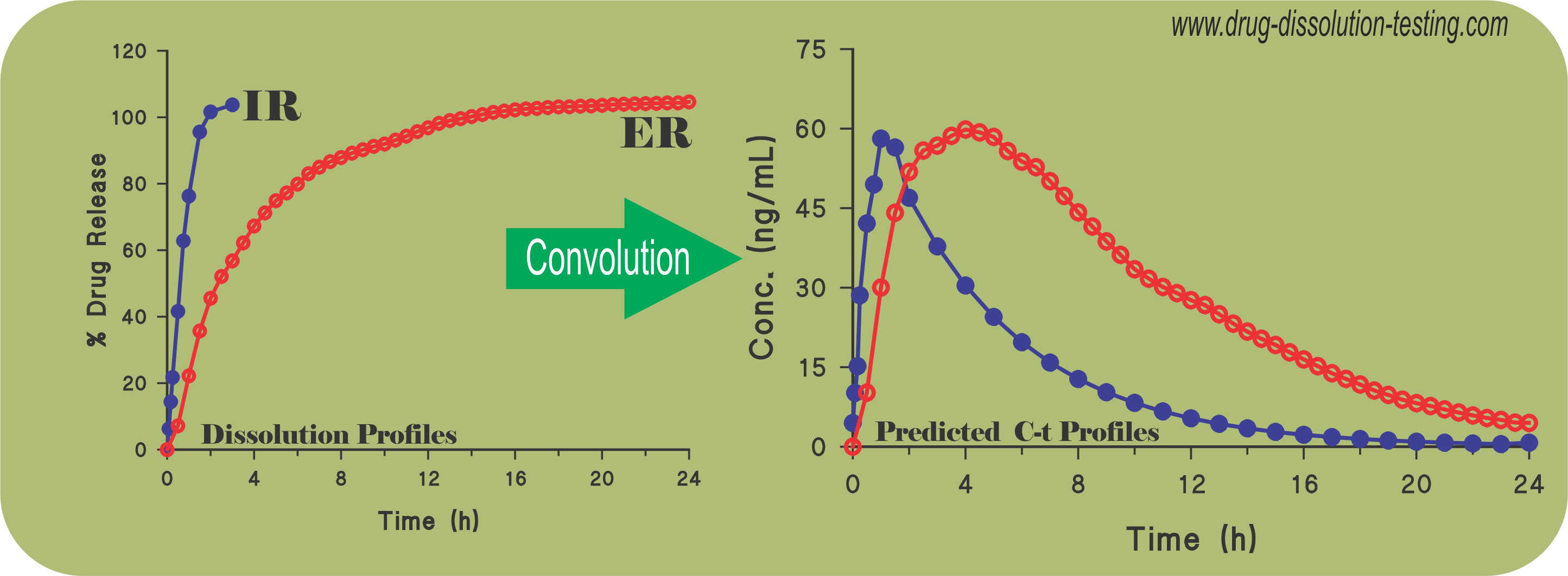
Recently a simple and practical approach has been suggested to predict plasma drug concentration-time profiles from drug dissolution results (link) without conducting a concurrent bio-study. The following citations describe application and usefulness of such an approach as reported in recent literature (refereed journals) from third-party laboratories.
Slides from a presentation made at the 2nd Annual Bioequivalence Conference: Intersection between Science and Regulatory Summit, Philadelphia, PA, USA. September 29-30, 2015 link.


{kind=link}