For further discussion, please see the following links (1, 2, 3)
It is a well-established understanding that the drug/pharmaceutical industry is highly regulated. The elaborate regulations’ purpose and implementation are that the products manufactured should be of the highest quality standards possible. There are numerous sources of such regulations, most often country- or territory-specific such as US, Japan, Europe, etc., or some others harmonized such as from ICH. However, the most commonly referred or quoted are those from the US FDA, US Pharmacopeia, EMA, and ICH.
Regulatory authorities such as US FDA, Health Canada, EMA, and many others enforce such regulations and standards to ascertain that manufacturers and manufactured products are in compliance with regulations leading to the manufacturing of quality products. It is very important to note that a fundamental underlying assumption here is that if a product or process is in compliance then the product or process will be of quality. In general, such an underlying assumption is correct; however, this underlying assumption does not appear to be valid for the pharmaceutical industry.
In general, regulations and standards are based on, or derived from, scientific research following underlying scientific principles and theories. A common terminology that is used to describe these underlying scientific principles and theories is “validation,” i.e., the process, or processes, has been validated to provide quality products. Every step (small or large) is considered or broken further into smaller steps to validate the result (or product) should be of quality. For example, analytical techniques (such as chromatographic instruments and methods) may not be directly considered a manufacturing step but are critical in monitoring the outcome of manufacturing, thus requiring validation of their own.
From the scientific and regulatory compliance perspectives, validation of manufacturing processes, along with associated steps or components, is perhaps the most important and critical step and/or requirement. It is further critical to note that regulatory compliance requirements are, or at least should be, dependent on the well-established validation steps. It is not practical or useful to develop and/or implement compliance requirements and/or standards without having validation steps first.
So what does a validation step/process mean? In simple terms, if a claim is made, it must be substantiated based on scientific (mostly experimental) evidence. For example, suppose it is claimed that a product is of quality. In that case, it becomes mandatory first to state what quality means and then how this defined quality is established using scientific methods. The regulatory mandate is to evaluate if the quality is defined accurately, and then the methods and processes used are capable of measuring the quality of the products. In general, regulatory standards and requirements focus primarily on the outcome of manufacturing and may not be manufacturing itself, which for all practical purposes is secondary in the assessment of the outcome (products).
From a regulatory perspective, one has to deal with two aspects; (1) what is a “quality” product or what is “quality” of a product, and (2) how is it measured or established experimentally (scientifically). Unfortunately, however, it is disturbing that the quality of a pharmaceutical product has never been defined, particularly for regulatory assessment purposes. Therefore, it is not possible, at present, to know or establish whether a, or any, given product is of quality even if the regulatory authorities approve it. This situation needs to be addressed and corrected on an urgent basis.
On the other hand, regulatory authorities suggest and enforce some traditional practices and standards, assuming that if manufacturers are in-compliance with such, the products would be considered of acceptable quality. Here again, there is a serious deficiency in suggesting compliance requirements. Often methods and techniques suggested, at least some, have never been validated for the claims made for them. For example, a technique known as drug dissolution testing, often mandatory for evaluating the quality of products, in particular oral such as tablet and capsule, has never been shown to provide any relevant characteristics of a product. There are so many stringent requirements for the required testers and testing methods without any scientific basis or reasoning. These are often extremely frustrating and resource-consuming exercises for the manufacturers and the regulatory authorities to meet compliance requirements for this test which has no link or contribution towards the quality assessment of the products. Recommendations and requirements for the use of non-validated and non-qualified testers and tests are serious violations of GMP requirements. Regulatory authorities should reconsider the current requirements of such testing on an urgent basis.
In short, as the quality of pharmaceutical products is an undefined parameter (metric), it is not possible that manufactured products can be assessed for quality. On the other hand, current regulatory requirements that manufacturers are to follow to comply with product approval are based on techniques and assumptions that lack scientific merit and/or validation of testers and methods, thus giving false hope or comfort about the quality of products.
The good news is that both of the issues mentioned above can be addressed with relative ease if a more logical and scientific approach/thinking would be pursued. I have extensively written about these issues and suggested possible solutions for addressing these issues by publishing in the scientific literature. Perhaps, the following articles would be useful as a start.
(1) Promoting quality standards for drug products: Scientifically speaking, please be systematic and logical! (link).
(2) Establishing safety, efficacy and quality of drugs and drug-products (tablet/capsule) – serious confusion! (link)
(3) The science of drug dissolution testing: Testers or apparatuses, experimental conditions and interpretation of results – A systematic approach for learning (link)
It is a well-established understanding that the drug/pharmaceutical industry is highly regulated. The elaborate regulations’ purpose and implementation are that the products manufactured should be of the highest quality standards possible. There are numerous sources of such regulations, most often country- or territory-specific such as US, Japan, Europe, etc., or some others harmonized such as from ICH. However, the most commonly referred or quoted are those from the US FDA, US Pharmacopeia, EMA, and ICH.
Regulatory authorities such as US FDA, Health Canada, EMA, and many others enforce such regulations and standards to ascertain that manufacturers and manufactured products are in compliance with regulations leading to the manufacturing of quality products. It is very important to note that a fundamental underlying assumption here is that if a product or process is in compliance then the product or process will be of quality. In general, such an underlying assumption is correct; however, this underlying assumption does not appear to be valid for the pharmaceutical industry.
In general, regulations and standards are based on, or derived from, scientific research following underlying scientific principles and theories. A common terminology that is used to describe these underlying scientific principles and theories is “validation,” i.e., the process, or processes, has been validated to provide quality products. Every step (small or large) is considered or broken further into smaller steps to validate the result (or product) should be of quality. For example, analytical techniques (such as chromatographic instruments and methods) may not be directly considered a manufacturing step but are critical in monitoring the outcome of manufacturing, thus requiring validation of their own.
From the scientific and regulatory compliance perspectives, validation of manufacturing processes, along with associated steps or components, is perhaps the most important and critical step and/or requirement. It is further critical to note that regulatory compliance requirements are, or at least should be, dependent on the well-established validation steps. It is not practical or useful to develop and/or implement compliance requirements and/or standards without having validation steps first.
So what does a validation step/process mean? In simple terms, if a claim is made, it must be substantiated based on scientific (mostly experimental) evidence. For example, suppose it is claimed that a product is of quality. In that case, it becomes mandatory first to state what quality means and then how this defined quality is established using scientific methods. The regulatory mandate is to evaluate if the quality is defined accurately, and then the methods and processes used are capable of measuring the quality of the products. In general, regulatory standards and requirements focus primarily on the outcome of manufacturing and may not be manufacturing itself, which for all practical purposes is secondary in the assessment of the outcome (products).
From a regulatory perspective, one has to deal with two aspects; (1) what is a “quality” product or what is “quality” of a product, and (2) how is it measured or established experimentally (scientifically). Unfortunately, however, it is disturbing that the quality of a pharmaceutical product has never been defined, particularly for regulatory assessment purposes. Therefore, it is not possible, at present, to know or establish whether a, or any, given product is of quality even if the regulatory authorities approve it. This situation needs to be addressed and corrected on an urgent basis.
On the other hand, regulatory authorities suggest and enforce some traditional practices and standards, assuming that if manufacturers are in-compliance with such, the products would be considered of acceptable quality. Here again, there is a serious deficiency in suggesting compliance requirements. Often methods and techniques suggested, at least some, have never been validated for the claims made for them. For example, a technique known as drug dissolution testing, often mandatory for evaluating the quality of products, in particular oral such as tablet and capsule, has never been shown to provide any relevant characteristics of a product. There are so many stringent requirements for the required testers and testing methods without any scientific basis or reasoning. These are often extremely frustrating and resource-consuming exercises for the manufacturers and the regulatory authorities to meet compliance requirements for this test which has no link or contribution towards the quality assessment of the products. Recommendations and requirements for the use of non-validated and non-qualified testers and tests are serious violations of GMP requirements. Regulatory authorities should reconsider the current requirements of such testing on an urgent basis.
In short, as the quality of pharmaceutical products is an undefined parameter (metric), it is not possible that manufactured products can be assessed for quality. On the other hand, current regulatory requirements that manufacturers are to follow to comply with product approval are based on techniques and assumptions that lack scientific merit and/or validation of testers and methods, thus giving false hope or comfort about the quality of products.
The good news is that both of the issues mentioned above can be addressed with relative ease if a more logical and scientific approach/thinking would be pursued. I have extensively written about these issues and suggested possible solutions for addressing these issues by publishing in the scientific literature. Perhaps, the following articles would be useful as a start.
(1) Promoting quality standards for drug products: Scientifically speaking, please be systematic and logical! (link).
(2) Establishing safety, efficacy and quality of drugs and drug-products (tablet/capsule) – serious confusion! (link)
(3) The science of drug dissolution testing: Testers or apparatuses, experimental conditions and interpretation of results – A systematic approach for learning (link)
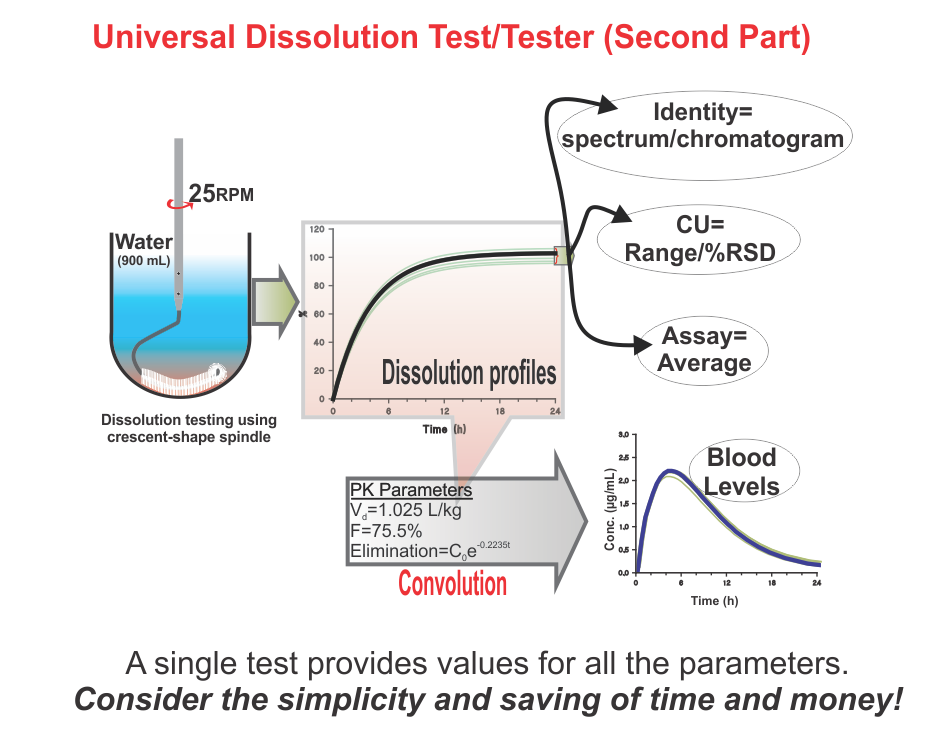
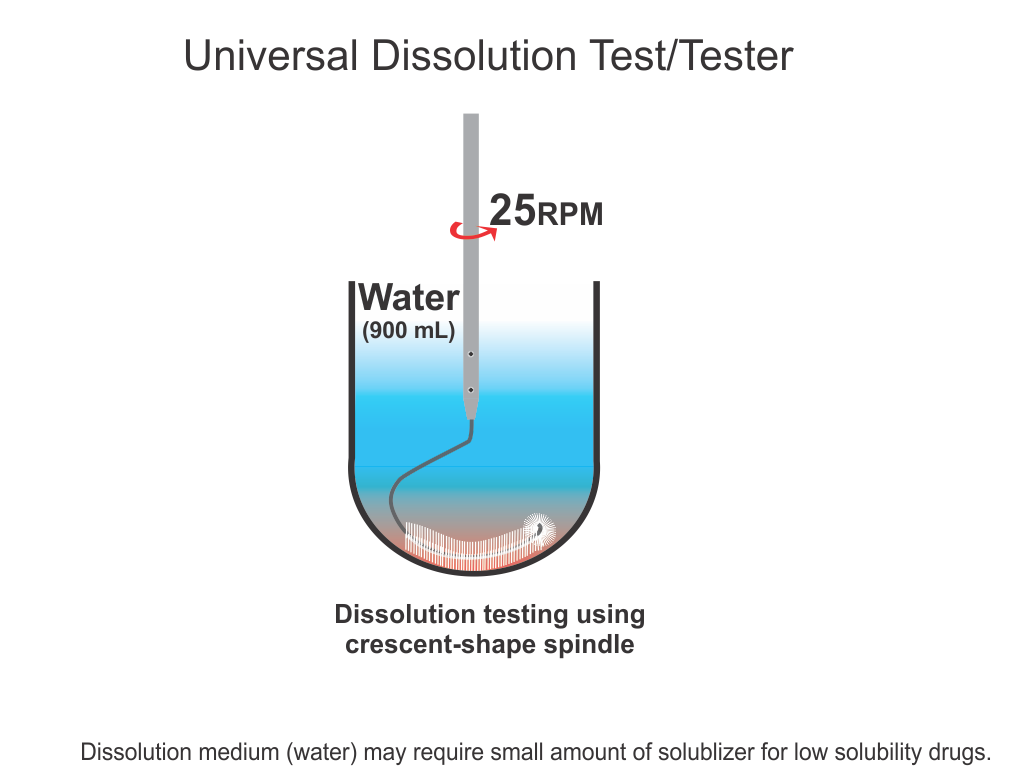
Bio-waiver is a term used for product approvals, particularly tablet/capsule, without requiring an in vivo (bio-equivalence/bio-availability) testing commonly required to establish the safety, efficacy, and quality of drug products. For bio-waivers, the requirement of bio-equivalence/bio-availability studies is substituted with in vitro drug release testing, commonly known as drug dissolution testing. It is, therefore, extremely important to note that drug dissolution testing is used as a surrogate of in vivo (bio-equivalence/bio-availability) testing. Stating otherwise is clearly not an accurate view or representation of the science.
In addition, regulatory, including pharmacopeial, requirements of in vitro drug dissolution testing are based on the assumption that if products met the dissolution test criteria, then products would be considered safe, efficacious, and of quality for human use. This is the reason that dissolution tests are often promoted as quality control/assurance tests for pharmaceutical products (e.g., tablets/capsules).
However, unfortunately, the dissolution tests (methods/testers) often recommended and used have never been qualified and/or validated for their intended purpose. The assessment of the quality of such products, using the recommended testers/methods, lacks scientific support and merit. Therefore, current practices of requiring bio-waivers lack scientific merit or credibility; thus, their requirements should be reconsidered.
As per a recent follow-up note from USP (link), it is stated that “It is known that the dissolution of USP Prednisone Tablets RS changes over time.” Is this a good thing?
In principle, if the tablets, or anything else, are not stable, then by default, these should not be used as a REFERENCE. How would their use as a REFERENCE be justified?
On the other hand, the new validity/expiry date has been extended for six months to December 31th, 2015. Will the tablets’ characteristics change gradually or will they change abruptly on December 31, 2015? If the dissolution characteristics’ changes are gradual, shouldn’t the Ranges be changed more frequently such as weekly, bi-weekly, or monthly, etc?
Some thoughts for consideration!
Last week USP once again, in a “surprise” announcement, informed that their current lot of Prednisone Tablets for PVT is not performing as expected, so its use is halted, at least for a while. The question is, is this a surprise or unusual occurrence? Anyone involved in drug dissolution testing, in particular PVT, would know that such occurrences are not unusual or new but becoming relatively frequent.
Anticipation is that USP might provide some information concerning the problem and hopefully the cause of such problems. However, past experiences show that such information may not be forthcoming; perhaps, some modifications to data presentation format and/or slight adjustments in Acceptance Ranges would be made with a claim that “all is well and good to go.”
Indeed, just today (a few days later from the previous announcement), it appears that the USP has released a continuation lot, with revised and adjusted Acceptance Ranges. Such scientifically poor practices from a reputed standard-setting organization such as USP are quite frustrating. USP should perhaps re-evaluate its practice of adjusting or re-adjusting the data “on the go”. Otherwise, manufacturers of the products should also be allowed to revise their products’ tolerances if their batches start giving different, or unexpected, sets of data!
On the other hand, the fact is that problems of PVT remain, i.e., frequent observance of PVT results falling outside the expected ranges reflecting high variability and unpredictability of the test. This high variability and lack of predictability in dissolution results are because of the testers (paddle and basket); however, unfortunately, USP Prednisone Tablets get the blame.
It is very important to note that to date, there has not been a single piece of evidence presented showing that indeed the failure of PVT is because of the Prednisone Tablets. On the other hand, many scientific studies have published showing that the high variability (or failures) is related to the dissolution testers (paddle and basket). Performance testing using Prednisone Tablets merely reflects the variability of the dissolution testers. It is just like if one’s computer monitor blacks out or flickers often, one has to assess separately whether the problem is associated with the monitor or the computer. If the computer is causing the problem, then adjusting or changing the monitor would not help. Similarly, in case of dissolution testing, adjusting or changing the Performance Tablets, and/or their Acceptance Ranges would not help. Therefore, USP must demonstrate that dissolution testers (paddle and basket) evaluated separately and independently from the Prednisone Tablets can provide predictable and relevant results i.e., these testers are validated and qualified to evaluate the dissolution characteristics of something or anything. Once this validation and qualification step has been accomplished, these testers should establish performance using Prednisone Tablets and/or actual drug products.
It’s hoped that the USP will use this latest miserable situation to address the well-known problem of the dissolution testers, not just by re-adjusting the Acceptance Ranges. Please consider providing a validated and qualified dissolution tester.