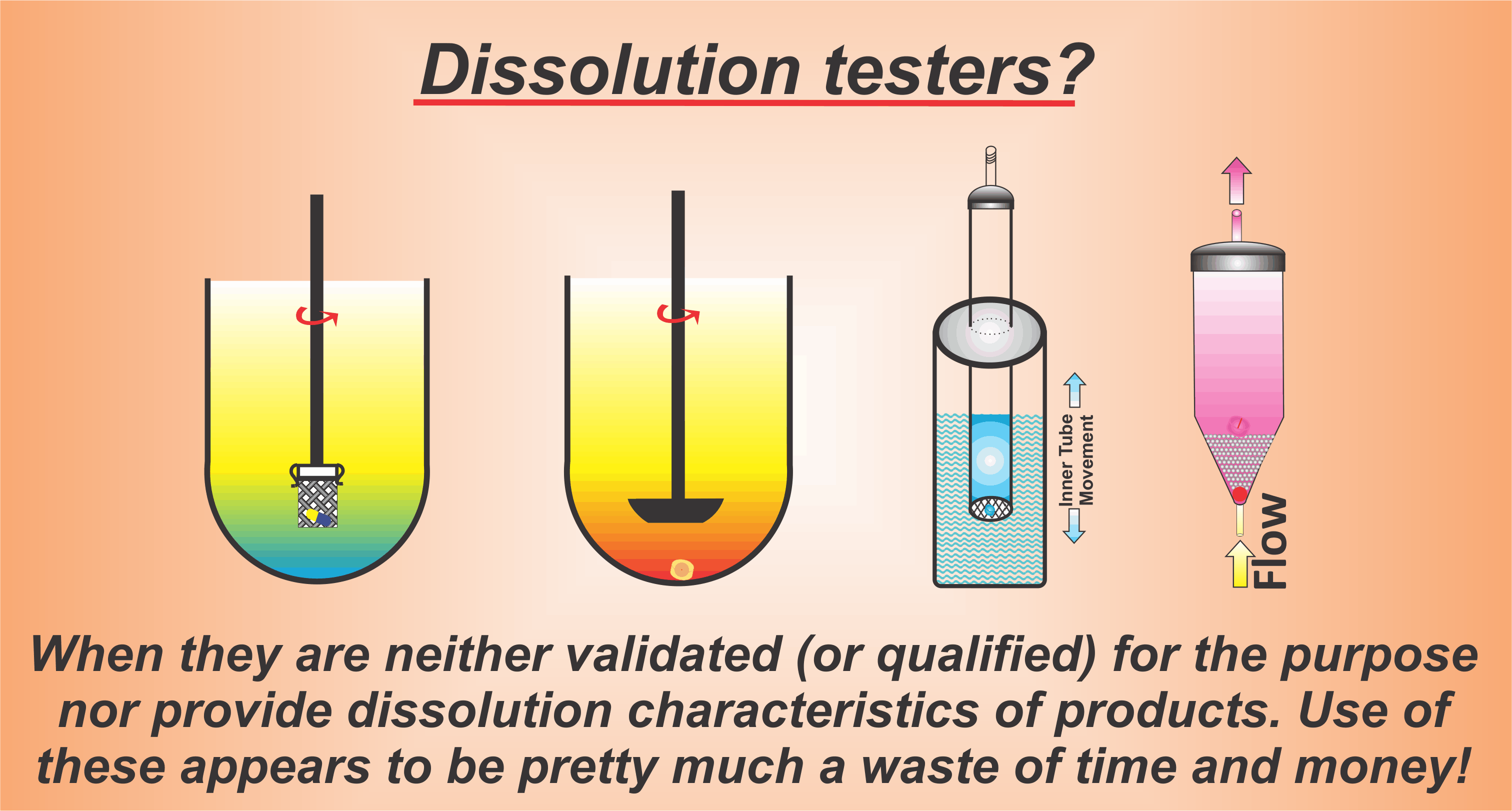
It is very important to note that the objective of dissolution testing is to estimate/determine potential drug dissolution/release characteristics of a drug from a product using a simulated gastrointestinal (GI) tract environment. For dissolution testing purposes, the simulated GI tract environment is commonly represented by a medium such as water or an aqueous buffer (e.g., pH=6) in a container (vessel) maintained at 37 °C with gentle mixing/stirring. Suppose the volume of the medium is not sufficient to completely dissolve the expected amount of drug present in the product. In that case, a small amount of solubilizer (e.g. SLS) may be used. Dissolution results are often expressed as a percentage of the drug released at a single time (e.g., 30 or 45 minutes) or multiple times to establish a dissolution profile.
A dissolution test is like any other simple analytical test utilizing an instrument, such as a thermometer, viscometer, or pH meter that one uses to measure temperature, viscosity, and pH of a product, respectively. One does not require or develop a product-specific thermometer, viscometer, or pH meter. One just puts the thermometer or electrode into the liquid or pours the liquid into the viscometer to obtain the reading. This reading will reflect the property of the liquid.
A dissolution tester should be used exactly like that, i.e., monitoring/measuring dissolution/release characteristics of a product (tablet/capsule) by dropping a tablet/capsule into a dissolution vessel containing a standard volume of dissolution medium with a stirrer a pre-set rpm. The outcome of the test (% dissolved) will reflect the dissolution characteristics of the product.
No matter how one presents the argument, or dissolution results, describing them such as bio- or IVIVC relevant, discriminatory, or for QC use, the tests must be performed using pre-set and product independent experimental conditions. This is similar to the practice of not requiring discriminatory or bio-relevant and/or liquid-dependent thermometers, viscometers, or pH meters. Not only will it logically and scientifically be considered meaningless but it will never reflect the true product’s characteristics. Similarly, one should not ask for and/or try to develop product-dependent dissolution testers or methods (Please, click here to read in detail about dissolution testing).
Dissolution testing using the crescent shape spindle has been developed based on these thoughts which provide simple, scientifically valid, and product-independent testing and evaluation. For further information in this regard, please see the following links (1, 2).