
Method development and validation are critical parts of drug dissolution testing and are required for an appropriate product evaluation. However, it is a common practice and often time confusing and frustrating exercise. In addition, these exercises are often described by different names such as developing QC/QA, bio-(relevant) and/or discriminatory methods. In addition, these method development exercises are further complicated by requiring different types for each drug and product. A single type of product of the same drug, in particular ER, can have multiple methods.
On the other hand, however, if the methods described in the literature are critically evaluated, it would be quite obvious that there are not significant differences in these methods. The majority of the methods described in the literature are based on USP apparatus 1 (Basket) or 2 (Paddle) set mostly at 100/75/50 rpm with water or some form of buffer, having a pH in the range of 5-7 as a medium. Usual method validation steps follow this experimental part. Confusion and frustration arise from the question of how to select a particular set of experimental conditions (apparatus, rpm, and medium). Unfortunately, there is no rationale or scientifically valid approach available for selecting the required set of experimental conditions. Formulators or analysts are often expected to make their own judgment calls, which causes the confusion and frustration.
{kind=link}

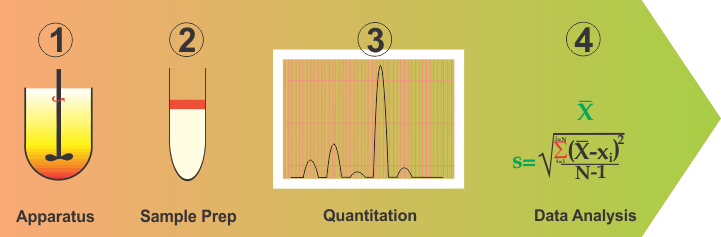
Although complex and difficult as this situation may be, one should be able to address this problem by simplifying and breaking down into parts the method development and validation exercises. In this regard, one can break this down into four distinct parts: (1) Apparatus containing medium; (2) Sampling or cleanup step; (3) Quantitation and; (4) Data analysis (or validation).
- An apparatus is a vessel with a stirrer containing some volume of water or buffer, representing the physiological environment within the GI tract, particularly of the small intestine. It is fairly easy to assume and construct such an environment. For most purposes, a one-liter vessel with a stirring rod containing water (with or without a solubilizing agent) serves well and has been in use for a long time. Let us assume that this arrangement reflects the required GI tract environment appropriately. If so, then one has already simplified the method development step because now all products should be tested using this environment and as the physiological environment does not change with the drugs and/or products, neither should be the apparatus and experimental conditions. For a more thorough discussion on this aspect, please see link .
- Data analysis (Method Validation) is basically an exercise of determining values of precision and accuracy parameters from the data/results. This is also common for all drugs and products.
- Quantitation: This is dependent on the drug and mostly represents the choice of a wavelength. This is often available from the literature or can easily be determined by a UV scan. However, it is important to note that it is not a dissolution issue as dissolution is conducted for a product while wavelength is a drug characteristic and is often known prior to the product development stage.
- Sampling or clean-up step. This clearly depends on the product in particular its excipients. This is where the expertise of an analyst is required to establish if the dissolution samples can be quantified as such or would require a clean-up step to remove interference if observed. Clean-up step may be off-line (simple liquid-liquid extraction) and/or on-line (chromatographic).
An important conclusion from the above discussion is that three are already set or fixed out of the four steps, and an analyst/chemist does not have to be concerned about them. It appears that dissolution method development and validation exercises boil down to step number two, i.e., determining an appropriate dissolution sample-clean up procedure.
The main reason for the confusion in the method development exercise is that analysts often work with step one, i.e., the apparatus part. They seek an appropriate apparatus with associated experimental conditions to evaluate their products. However, realistically this practice is incorrect, as it assumes that the GI tract environment will be different or linked to their product. The fact is that the GI tract remains the same and is not product-dependent.
On the other hand, if the intent is to develop a new apparatus and/or its associated experimental conditions, or improve upon a currently employed approach, then this practice should be considered as an apparatus and/or experimental conditions development step, not a “method” development step. Apparatus/experimental conditions development is very different and requires an analyst to demonstrate that the suggested apparatus and associated test conditions do reflect an improved GI tract environment. In addition, such development will require a “product” with known and well-recognized dissolution characteristics. Such an approach cannot be used for product characterization or evaluation.
Therefore, in short, the current practice of “method development” only requires development and validation of the sample prep step. Changing the apparatus and/or associated experimental conditions for a product under development would invalidate a dissolution method and its results.